createIndices
What it does
This is a special pipeline in that it creates index files required by various tools within snakePipes. This workflow takes as input a fasta file (or URL) and GTF file (or URL) as well as various optional files and generates both indices and the organism yaml file used by snakePipes.
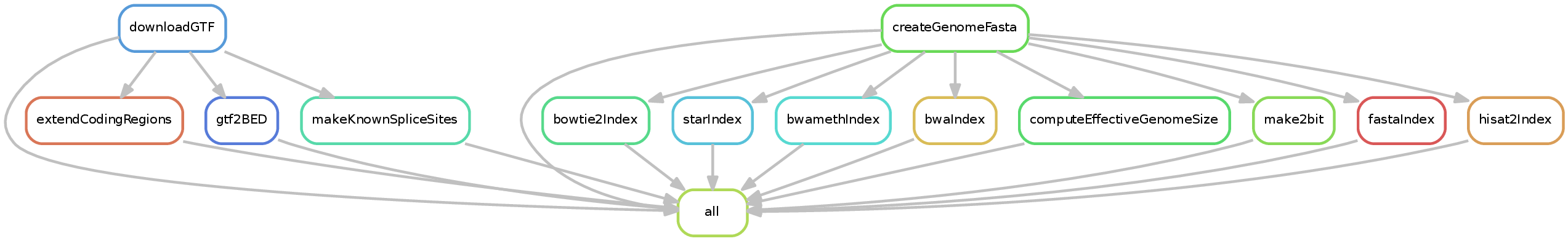
Input requirements
The pipeline has two required inputs: a fasta file or URL and a GTF file or URL. These may both be gzipped. Optionally, you may specify a blacklist file (such as that provided by ENCODE), an effective genome size, and a file listing chromosomes to be ignored during normalization steps.
Note
If you specify a blacklist file, please ensure that regions within it do NOT overlap. Overlapping regions in this file will cause incorrect results in some tools. Further, it is best to flank blacklisted regions by at least 50 bases, as otherwise many reads originating within these regions may be nonetheless included.
Configuration file
There is a configuration file in snakePipes/workflows/createIndices/defaults.yaml:
pipeline: createIndices
outdir:
configFile:
clusterConfigFile:
local: False
maxJobs: 5
verbose: False
## Genome name used in snakePipes (no spaces!)
genome:
## Tools to create indices for. "all" for all of them
tools: all
## URLs or paths for fasta and GTF files
genomeURL:
spikeinGenomeURL:
spikeinExt: '_spikein'
gtfURL:
spikeinGtfURL:
## The effective genome size
effectiveGenomeSize: 0
## Regions to blacklist in the ChIPseq and related workflows
blacklist:
spikeinBlacklist:
## Regions to ignore during normalization (e.g., with bamCompare)
ignoreForNormalization:
## Repeat masker file. It's assumed that the columns are tab separated!
rmsk_file:
## Salmon Index Options
salmonIndexOptions: --type puff -k 31
eisaR_flank_length: 80
These values are most conveniently set on the command line.
Hybrid genome
To create a hybrid fasta, specify the host genome with --genomeURL and the spikein genome with --spikeinGenomeURL. On top of --gtfURL and --blacklist, you may optionally provide --spikeinGtfURL and --spikeinBlacklist. Default extention added to spikein chromosomes is '_spikein' and can be changes with --spikeinExt.
Output structure
The following structure will be created in the designated outdir:
.
├── annotation
├── BowtieIndex
├── BWAIndex
├── BWAmethIndex
├── createIndices.cluster_config.yaml
├── createIndices.config.yaml
├── createIndices_run-1.log
├── genome_fasta
├── HISAT2Index
├── STARIndex
├── SalmonIndex
└── SalmonIndex_RNAVelocity
These files are used internally within snakePipes and don't require further inspection. The createIndices_run-1.log file contains a full log and will include the URLs or file paths that you specified. Whether the annotation/blacklist.bed file exists is dependent upon whether you specified one. The genome_fasta/effectiveSize fill will have the effective genome size (if you didn't specify it, the number of non-N bases in the genome will be used).
In addition to these, an organism yaml file will be created. Its location can be found with snakePipes info.
Command line options
Create indices for use by snakePipes. A YAML file will be created by default in the default location where snakePipes looks for organism YAML files.
- usage example:
createIndices -o output-dir --genome ftp://ftp.ensembl.org/pub/release-93/fasta/mus_musculus/dna/Mus_musculus.GRCm38.dna_sm.primary_assembly.fa.gz --gtf ftp://ftp.ensembl.org/pub/release-93/gtf/mus_musculus/Mus_musculus.GRCm38.93.gtf.gz --blacklist blacklist.bed --ignoreForNormalization ignore.txt GRCm38_release93
usage: createIndices -o OUTDIR [-h] [-v] [-c CONFIGFILE] [--keepTemp]
[--snakemakeOptions SNAKEMAKEOPTIONS] [--DAG] [--version]
[--emailAddress EMAILADDRESS] [--smtpServer SMTPSERVER]
[--smtpPort SMTPPORT] [--onlySSL]
[--emailSender EMAILSENDER] [--smtpUsername SMTPUSERNAME]
[--smtpPassword SMTPPASSWORD] --genomeURL GENOMEURL
[--gtfURL GTFURL] [--spikeinGenomeURL SPIKEINGENOMEURL]
[--spikeinGtfURL SPIKEINGTFURL] [--spikeinExt SPIKEINEXT]
[--tools {all,bowtie2,hisat2,bwa,bwa-mem2,bwameth,bwameth2,salmon,star,none} [{all,bowtie2,hisat2,bwa,bwa-mem2,bwameth,bwameth2,salmon,star,none} ...]]
[--effectiveGenomeSize EFFECTIVEGENOMESIZE]
[--spikeinBlacklist SPIKEINBLACKLIST]
[--blacklist BLACKLIST]
[--ignoreForNormalization IGNOREFORNORMALIZATION]
[--rmskURL RMSKURL]
[--salmonIndexOptions SALMONINDEXOPTIONS]
[--eisaR_flank_length EISAR_FLANK_LENGTH]
GENOME
Positional Arguments
- GENOME
The name to save this genome as. No spaces or special characters! Specifying an organism that already exists will cause the old information to be overwritten. See also the --userYAML option.
Required Arguments
- -o, --output-dir
output directory
- --genomeURL
URL or local path to where the genome fasta file is located. The file may optionally be gzipped.
- --gtfURL
URL or local path to where the genome annotation in GTF format is located. GFF is NOT supported. The file may optionally be gzipped. If this file is not specified, then RNAseq related tools will NOT be usable.
General Arguments
- -v, --verbose
verbose output (default: 'False')
- -c, --configFile
configuration file: config.yaml (default: 'None')
- --keepTemp
Prevent snakemake from removing files marked as being temporary (typically intermediate files that are rarely needed by end users). This is mostly useful for debugging problems.
- --snakemakeOptions
Snakemake options to be passed directly to snakemake, e.g. use --snakemakeOptions='--dryrun --rerun-incomplete --unlock --forceall'. WARNING! ONLY EXPERT USERS SHOULD CHANGE THIS! THE DEFAULT VALUE WILL BE APPENDED RATHER THAN OVERWRITTEN! (default: '['']')
- --DAG
If specified, a file ending in _pipeline.pdf is produced in the output directory that shows the rules used and their relationship to each other.
- --version
show program's version number and exit
Email Arguments
- --emailAddress
If specified, send an email upon completion to the given email address
- --smtpServer
If specified, the email server to use.
- --smtpPort
The port on the SMTP server to connect to. A value of 0 specifies the default port.
- --onlySSL
The SMTP server requires an SSL connection from the beginning.
- --emailSender
The address of the email sender. If not specified, it will be the address indicated by --emailAddress
- --smtpUsername
If your SMTP server requires authentication, this is the username to use.
- --smtpPassword
If your SMTP server requires authentication, this is the password to use.
Options
- --spikeinGenomeURL
URL or local path to where the spikein genome fasta file is located. The file may optionally be gzipped.
- --spikeinGtfURL
URL or local path to where the spikein genome annotation in GTF format is located. GFF is NOT supported. The file may optionally be gzipped.
- --spikeinExt
Extention of spikein chromosome names in the hybrid genome. (default: 'None') .
- --tools
Possible choices: all, bowtie2, hisat2, bwa, bwa-mem2, bwameth, bwameth2, salmon, star, none
Only produce indices for the following tools (by default, all indices will be created). The default is 'all'. 'none' will create everything except aligner indices.
- --effectiveGenomeSize
The effective genome size. If you don't specify a value then the number of non-N bases will be used.
- --spikeinBlacklist
An optional URL or local path to a file to use to blacklist spikein organism regions (such as that provided by the ENCODE consortium).
- --blacklist
An optional URL or local path to a file to use to blacklist regions (such as that provided by the ENCODE consortium).
- --ignoreForNormalization
An optional file list, with one entry per line, the chromosomes to ignore during normalization. These are typically sex chromosomes, mitochondrial DNA, and unplaced contigs.
- --rmskURL
URL or local path to where the repeat masker output file is located. This is only required if you plan to run the ncRNAseq workflow.
- --salmonIndexOptions
Options to pass to salmon for index creation.
- --eisaR_flank_length
Length by which to extend intronic regions with eisaR.