HiC
What it does
The snakePipes HiC workflow allows users to process their HiC data from raw fastq files to corrected HiC matrices and TADs. The workflow utilized mapping by bwa or bwa-mem2 , followed by analysis using HiCExplorer. The workflow follows the example workflow described in the documentation of HiCExplorer, which explains each step in detail and would be useful for new users to have a look at. The output matrices are produced in the .hdf5 format.
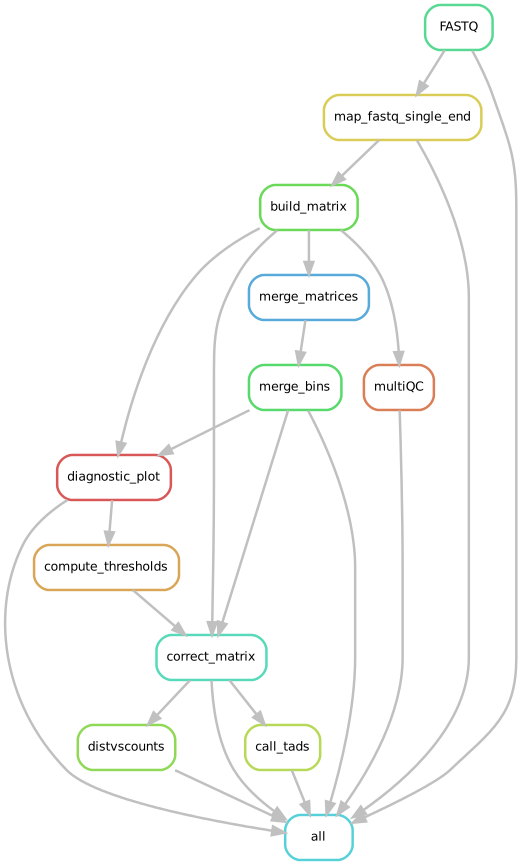
Input requirements and outputs
This pipeline requires paired-end reads fastq files as input in order to build a contact matrix and to call TADs. Prior to building the matrix, the pipeline maps reads against a user-specified reference genome. The output of mapping step is then used for building the contact matrix.
Workflow configuration file
Default parameters from the provided config file can be altered by user. Below is the config file description for the HiC workflow :
################################################################################
# This file is the default configuration of the HiC workflow!
#
# In order to adjust some parameters, please either use the wrapper script
# (eg. /path/to/snakemake_workflows/workflows/HiC/HiC)
# or save a copy of this file, modify necessary parameters and then provide
# this file to the wrapper or snakmake via '--configFile' option
# (see below how to call the snakefile directly)
#
# Own parameters will be loaded during snakefile executiuon as well and hence
# can be used in new/extended snakemake rules!
################################################################################
## General/Snakemake parameters, only used/set by wrapper or in Snakemake cmdl, but not in Snakefile
pipeline: hic
outdir:
configFile:
clusterConfigFile:
#set to true if running locally
local: False
#number of threads
maxJobs: 5
## directory with fastq files
indir:
## preconfigured target genomes (mm9,mm10,dm3,...) , see /path/to/snakemake_workflows/shared/organisms/
## Value can be also path to your own genome config file!
genome:
## FASTQ file extension (default: ".fastq.gz")
ext: '.fastq.gz'
## paired-end read name extension (default: ['_R1', "_R2"])
reads: ['_R1', '_R2']
aligner: BWA
## Number of reads to downsample from each FASTQ file
downsample:
## Options for trimming and fastqc
trim: False
trimmer: cutadapt
trimmerOptions:
fastqc: false
verbose: False
## is the Matrix RF resolution?
RFResolution: false
# which restriction enzyme was used
enzymes: HindIII
# bin size in base pairs, if RF resolution is not required
binSize: 10000
# build matrix only for a given region chr:start-end
restrictRegion:
# Create files after merging a given number bins to be merged (default 0 = bins are not merged)
nBinsToMerge: 0
# shall we merge the samples?
mergeSamples: false
# parameters for hicFindTADs
findTADParams: '--thresholdComparisons 0.01'
# Should hicPlotDistVsCounts be run?
distVsCount: false
#Parameters to run hicPlotDistVsCounts
distVsCountParams:
#Terminate the pipeline before calling TADs
noTAD: false
#Terminate the pipeline before correting the matrices with a certain cutoff value
noCorrect: false
# Method to balance a matrix
correctionMethod: KR
#Chromosomes of interest to build matrix on them
chromosomes:
# a .tsv file contains names and replicates of samples. It is needed if mergeSamples
sampleSheet:
#print tools versions
toolsVersion: True
#umi_tools
UMIBarcode: False
bcPattern: NNNNCCCCCCCCC #default: 4 base umi barcode, 9 base cell barcode (eg. RELACS barcode)
UMIDedup: False
UMIDedupSep: "_"
UMIDedupOpts: --paired
################################################################################
# Call snakemake directly, i.e. without using the wrapper script:
#
# Please save a copy of this config yaml file and provide an adjusted config
# via '--configFile' parameter!
# example call:
#
# snakemake --snakefile /path/to/snakemake_workflows/workflows/HiC/Snakefile
# --configFile /path/to/snakemake_workflows/workflows/HiC/defaults.yaml
# --directory /path/to/outputdir
# --cores 32
################################################################################
Structure of output directory
In addition to the FASTQ module results (see Running snakePipes), the workflow produces the following outputs:
.
|--bwa
|--FASTQ
|--HiC_matrices
| |--logs
| |--QCplots
|--HiC_matrices_corrected
| |--logs
|--TADs
|--logs
bwa folder contains the mapping results in BAM format. The files were obtained after running bwa on each of the paired-end reads individually.
HiC_matrices folder accommodates the contact matrices generated by hicBuildMatrix. In case of merging samples or merging bins the initial matrix is saved in this folder along with the merged ones.
QCplot includes the QC measurements for each sample along with a diagnostic plot which illustrates a distribution of counts per bin. This information can be used to set a cutoff to prune (correct) the contact matrix.
Note
The cutoff value is computed by the pipeline and by default will be applied to build a corrected matrix. Generated matrices by the pipeline can further be used for downstream analysis such as detecting A/B compartments and they can also be visualized using hicPlotMatrix.
HiC_matrices_corrected folder is in fact containing the corrected matrix which has been generated via hicCorrectMatrix after pruning as has been mentioned above.
TADs folder includes the output of calling TADs using hicFindTADs. The output contains TAD boundaries, TAD domains and TAD scores. These along with the matrices can be visualized together as several tracks using pyGenomeTracks or can be interactively browsed via hicBrowser. Check figure below as an example.

Command line options
MPI-IE workflow for Hi-C analysis
- usage example:
HiC -i input-dir -o output-dir mm10
usage: HiC -i INDIR -o OUTDIR [-h] [-v] [--ext EXT] [--reads READS READS]
[-c CONFIGFILE] [--keepTemp] [--snakemakeOptions SNAKEMAKEOPTIONS]
[--DAG] [--version] [--emailAddress EMAILADDRESS]
[--smtpServer SMTPSERVER] [--smtpPort SMTPPORT] [--onlySSL]
[--emailSender EMAILSENDER] [--smtpUsername SMTPUSERNAME]
[--smtpPassword SMTPPASSWORD] [--downsample INT] [--trim]
[--trimmer {cutadapt,trimgalore,fastp}]
[--trimmerOptions TRIMMEROPTIONS] [--fastqc] [--bcExtract]
[--bcPattern BCPATTERN] [--UMIDedup] [--UMIDedupSep UMIDEDUPSEP]
[--UMIDedupOpts UMIDEDUPOPTS] [--aligner {bwa,bwa-mem2}]
[--RFResolution] [--enzyme {DpnII,HindIII,MseI,CviQI}]
[--binSize INT] [--restrictRegion STR] [--mergeSamples]
[--nBinsToMerge NBINSTOMERGE] [--findTADParams STR] [--noTAD]
[--noCorrect] [--distVsCount] [--distVsCountParams STR]
[--sampleSheet STR] [--correctionMethod STR]
GENOME
Positional Arguments
- GENOME
Genome acronym of the target organism. Either a yaml file or one of: mm39_ens106, GRCz11, SchizoSPombe_ASM294v2, GRCh38_gencode40, hg38, GRCz10, mm10_gencodeM19, dm6
Required Arguments
- -i, --input-dir
input directory containing the FASTQ files, either paired-end OR single-end data
- -o, --output-dir
output directory
General Arguments
- -v, --verbose
verbose output (default: 'False')
- --ext
Suffix used by input fastq files (default: ''.fastq.gz'').
- --reads
Suffix used to denote reads 1 and 2 for paired-end data. This should typically be either '_1' '_2' or '_R1' '_R2' (default: '['_R1', '_R2']). Note that you should NOT separate the values by a comma (use a space) or enclose them in brackets.
- -c, --configFile
configuration file: config.yaml (default: 'None')
- --keepTemp
Prevent snakemake from removing files marked as being temporary (typically intermediate files that are rarely needed by end users). This is mostly useful for debugging problems.
- --snakemakeOptions
Snakemake options to be passed directly to snakemake, e.g. use --snakemakeOptions='--dryrun --rerun-incomplete --unlock --forceall'. WARNING! ONLY EXPERT USERS SHOULD CHANGE THIS! THE DEFAULT VALUE WILL BE APPENDED RATHER THAN OVERWRITTEN! (default: '['--use-conda']')
- --DAG
If specified, a file ending in _pipeline.pdf is produced in the output directory that shows the rules used and their relationship to each other.
- --version
show program's version number and exit
Email Arguments
- --emailAddress
If specified, send an email upon completion to the given email address
- --smtpServer
If specified, the email server to use.
- --smtpPort
The port on the SMTP server to connect to. A value of 0 specifies the default port.
- --onlySSL
The SMTP server requires an SSL connection from the beginning.
- --emailSender
The address of the email sender. If not specified, it will be the address indicated by --emailAddress
- --smtpUsername
If your SMTP server requires authentication, this is the username to use.
- --smtpPassword
If your SMTP server requires authentication, this is the password to use.
Options
- --downsample
Downsample the given number of reads randomly from of each FASTQ file (default: 'False')
- --trim
Activate fastq read trimming. If activated, Illumina adaptors are trimmed by default. Additional parameters can be specified under --trimmerOptions. (default: 'False')
- --trimmer
Possible choices: cutadapt, trimgalore, fastp
Trimming program to use: Cutadapt, TrimGalore, or fastp. Note that if you change this you may need to change --trimmerOptions to match! (default: ''cutadapt'')
- --trimmerOptions
Additional option string for trimming program of choice. (default: '''')
- --fastqc
Run FastQC read quality control (default: 'False')
- --bcExtract
To extract umi barcode from fastq file via UMI-tools and add it to the read name (default: 'False')
- --bcPattern
The pattern to be considered for the barcode. 'N' = UMI position (required) 'C' = barcode position (optional) (default: ''NNNNCCCCCCCCC'')
- --UMIDedup
Deduplicate bam file based on UMIs via umi_tools dedup that are present in the read name. (default: 'False')
- --UMIDedupSep
umi separation character that will be passed to umi_tools.(default: ''_'')
- --UMIDedupOpts
Additional options that will be passed to umi_tools.(default: '''')
- --aligner
Possible choices: bwa, bwa-mem2
Program used for mapping: bwa or bwa-mem2 (default: 'None').
- --RFResolution
Create Hi-C matrices at the restriction fragment resolution. Using RFResolution would override the --binSize argument. (default: 'False')
- --enzyme
Possible choices: DpnII, HindIII, MseI, CviQI
Which enzyme was used to create Hi-C library. Invoke multiple times if more than one enzyme was used (default: '['HindIII']')
- --binSize
Create Hi-C matrices at the given binSize. This option is mutally exclusive with the --RFResolution option (default: '10000')
- --restrictRegion
Restrict building of HiC Matrix to given region [Chr:Start-End]. Only one chromosome can also be specified (default: 'None')
- --mergeSamples
Merge HiC matrices and create a new matrix. If this option is specified togather with --sampleInfo (see below), the samples would be merged based on the defined groups. (default: 'False')
- --nBinsToMerge
If > 0 , create a lower resolution HiC matrix for each sample by merging the given number of bins. (default: '0')
- --findTADParams
parameters for HiCFindTADs. (default: ''--thresholdComparisons 0.01'')
- --noTAD
Stop the pipeline before TAD calling. (default: 'False')
- --noCorrect
Stop the pipeline before ICE-correction (i.e. run only upto building the matrix). (default: 'False')
- --distVsCount
Produce a plot of the ICE-corrected HiC counts as a function of distance. This plot could be used for QC as well as comparison between samples for biological effects. The plot is create using the tool 'hicDistVsCount'. (default: 'False')
- --distVsCountParams
parameters to run hicDistVsCount. (default: 'None')
- --sampleSheet
A .tsv file containing sample names and associated groups. If provided, the file would be used to identify groups to merge the samples. An example can be found at 'docs/content/sampleSheet.example.tsv'(default: None)
- --correctionMethod
Method to be used to balance the hic matrix. Available options are KR and ICE. (default: ''KR'')