mRNA-seq¶
What it does¶
The snakePipes mRNA-seq workflow allows users to process their single or paired-end mRNA-seq fastq files upto the point of gene/transcript-counts and differential expression. It also allows full allele-specific mRNA-seq analysis (up to allele-specific differential expression) using the allelic-mapping mode.
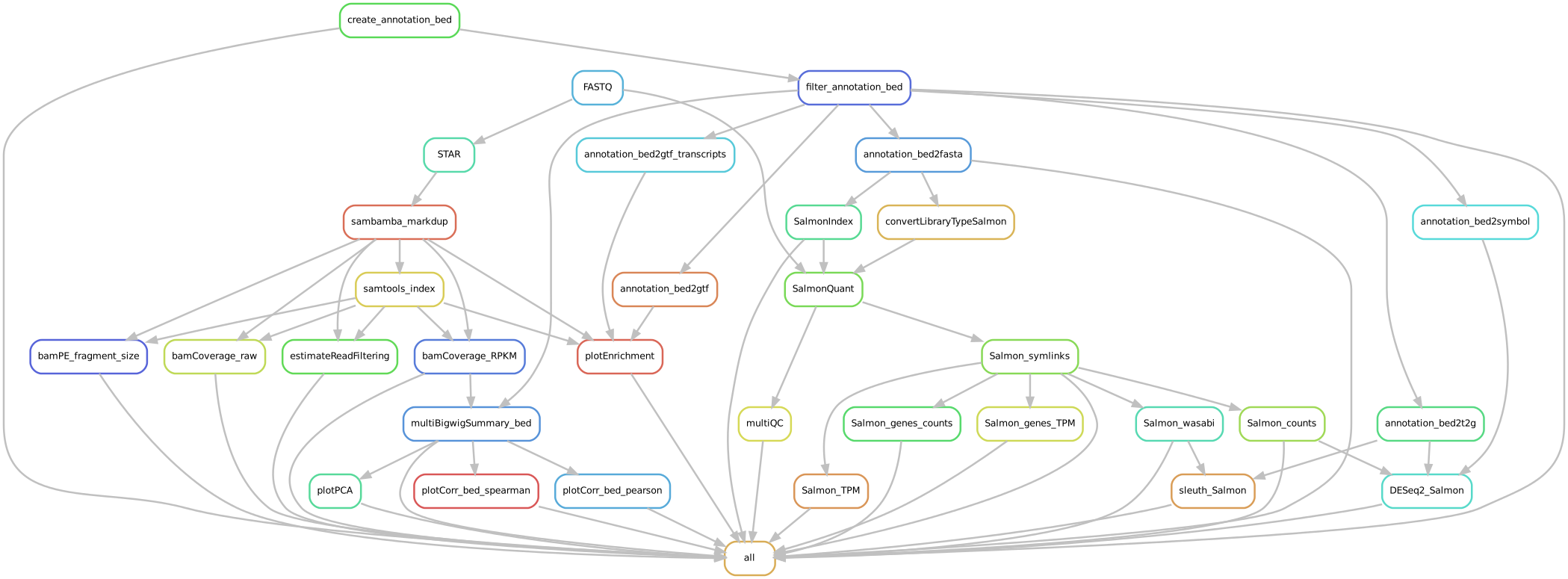
Input requirements¶
The only requirement is a directory of gzipped fastq files. Files could be single or paired end, and the read extensions could be modified using the keys in the defaults.yaml file below.
Configuration file¶
There is a configuration file in snakePipes/workflows/mRNA-seq/defaults.yaml:
pipeline: rna-seq
outdir:
configFile:
clusterConfigFile:
local: False
maxJobs: 5
## directory with fastq files
indir:
## preconfigured target genomes (mm9,mm10,dm3,...) , see /path/to/snakemake_workflows/shared/organisms/
## Value can be also path to your own genome config file!
genome:
## FASTQ file extension (default: ".fastq.gz")
ext: '.fastq.gz'
## paired-end read name extension (default: ["_R1", "_R2"])
reads: ["_R1","_R2"]
## assume paired end reads
pairedEnd: True
## Number of reads to downsample from each FASTQ file
downsample:
## Options for trimming
trim: False
trimmer: cutadapt
trimmerOptions:
## three prime seq options
# fastp options for three prime sequencing only
threePrimeTrimmerOptions: -x --poly_x_min_len 6 -3 5 -q 5 -l 20 -y
# STAR fastp options for three prime sequencing only
threePrimeAlignerOptions: --limitBAMsortRAM 60000000000 --alignIntronMax 1UMIDedupOpts: --paired
# parameters for calling polyA/T stretches in three_prime_seq workflow
polyAT:
minlength: 6
mindistance: 25 # recommendation for Drosophila; for other genomes might be different
extend: 3 # recommendation from Andrew
windowlength: 10
percbase: 0.7
# parameters for geneAssociation rule in three_prime_seq workflow
geneAssociation:
extend: 500
# parameters for cmatrix_raw in three_prime_seq workflow
cmatrix_raw:
upstream: 500
downstream: 500
clusterPAS:
window: 15
## further options
mode: alignment,deepTools_qc
sampleSheet:
rMats: False
bwBinSize: 25
fastqc: False
featureCountsOptions: -C -Q 10 --primary
filterGTF:
fragmentLength: 200
libraryType: 2
dnaContam: False
## supported mappers: STAR HISAT2
aligner: STAR
alignerOptions:
verbose: False
plotFormat: png
# for allele-spcific mapping
SNPFile:
NMaskedIndex:
#### Flag to control the pipeline entry point
fromBAM: False
bamExt: '.bam'
#umi_tools
UMIBarcode: False
bcPattern: NNNNCCCCCCCCC #default: 4 base umi barcode, 9 base cell barcode (eg. RELACS barcode)
UMIDedup: False
UMIDedupSep: "_"
UMIDedupOpts: --paired
Apart from the common workflow options (see Running snakePipes), the following parameters are useful to consider:
aligner: You can choose either STAR or HISAT2. While HISAT2 requires less memory than STAR, we keep STAR as the default aligner due to its superior accuracy (see this paper). Make sure that
--alignerOptionsmatches this.alignerOptions: Options to pass on to your chosen aligner. Note that library type and junction definitions doesn't have to be passed to the aligners as options, as they are handeled either automatically, or via other parameters.
featureCountsOptions: Options to pass to featureCounts (in case the
alignmentorallelic-mappingmode is used). Note that the paired-end information is automatically passed to featurecounts via the workflow, and the summerization is always performed at gene level, since the workflow assumes that featurecounts output is being used for gene-level differential expression analysis. If you wish to perform a transcript-level DE analysis, please choose the mode alignment-free.filterGTF: Options you can pass on to filter the original GTF file. This is useful in case you want to filter certain kind of transcripts (such as pseudogenes) before running the counts/DE analysis.
libraryType: The default library-type is suitable for most RNAseq protocols (using Illumina Tru-Seq). Change this option in case you have a different strandednes.
salmonIndexOptions: In the
alignment-freemode (see below), this option allows you to change the type of index created by salmon. New users can leave it to default.dnaContam: Enable this to test for possible DNA contamination in your mRNA-seq samples. DNA contamination is quantified as the fraction of reads falling into intronic and intergenic regions, compared to those falling into exons. Enabling this option would produce a directory called
GenomicContaminationwith.tsvfiles containing this information.plotFormat: You can switch the type of plot produced by all deeptools modules using this option. Possible choices : png, pdf, svg, eps, plotly
SNPFile: For the
allelic-mappingmode. TheSNPFileis the file produced by SNPsplit after creating a dual-hybrid genome. The file has the suffix.vcf.NMaskedIndex: For the
allelic-mappingmode. TheNMaskedIndexrefers to the basename of the index file created using STAR, on the SNPsplit output.
Note
SNPFile and NMaskedIndex file could be specified in case you already have this and would like to re-run the analysis on new data. In case you are running the allele-specific analysis for the first time, you would need a VCF file and the name of the two strains. In this case the SNPFile as well as the NMaskedIndex files would be automatically created by the workflow using SNPsplit.
Differential expression¶
Like the other workflows, differential expression can be performed using the --sampleSheet option and supplying a sample sheet like that below:
name condition
sample1 eworo
sample2 eworo
SRR7013047 eworo
SRR7013048 OreR
SRR7013049 OreR
SRR7013050 OreR
Note
The first entry defines which group of samples are control. This way, the order of comparison and likewise the sign of values can be changed. The DE analysis might fail if your sample names begin with a number. So watch out for that!
Optionally, the user may submit their desired model formula (without the leading tilda ~) with --formula.
Differential Splicing¶
In addition to differential expression, differential splicing analysis can be performed by using --rMats option in addition to supplying a sample sheet. This will invoke the rMats turbo on the samples.
Complex designs with blocking factors¶
If the user provides additional columns between 'name' and 'condition' in the sample sheet, the variables stored there will be used as blocking factors in the order they appear in the sample sheet. Eg. if the first line of your sample sheet looks like 'name batch condition', this will translate into a formula batch + condition. 'condition' has to be the final column and it will be used for any statistical inference.
Multiple pairwise comparisons¶
The user may specify multiple groups of independent comparisons by providing a 'group' column after the 'condition' column. This will cause the sample sheet to be split into the groups defined in this column, and a corresponding number of independent pairwise comparisons will be run, one for each split sheet, and placed in separate output folders named accordingly. This will be applied to DESeq2, sleuth, and rMats pairwise comparisons as requested by the user. Specifying a value of 'All' in the 'group' column will cause that sample group to be used in all pairwise comparisons, e.g. if the same set of controls should be used for several different treatment groups.
An example sample sheet with the group information provided looks like this:
name condition group sample1 Control All sample2 Control All sample3 Treatment Group1 sample4 Treatment Group1 sample5 Treatment Group2 sample6 Treatment Group2
Analysis modes¶
Following analysis (modes) are possible using the mRNA-seq workflow:
"alignment"¶
In this mode, the pipeline uses one of the selected aligners to create BAM files, followed by gene-level quantification using featureCounts. Gene-level differential expression analysis is then performed using DESeq2.
"allelic-mapping"¶
allelic-mapping mode follows a similar process as the "mapping" mode, however the alignment performed on an allele-masked genome, followed by allele-specific splitting of mapped files. Gene-level quantification is performed for each allele using featureCounts. Allele-specific, gene-level differential expression analysis is then performed using DESeq2.
Note
allelic-mapping mode is mutually exclusive with mapping mode
"allelic-counting"¶
allelic-counting mode requires the user to input, per sample, 4 bam files, corresponding to haplotype1, haplotype2, unassigned and haplotagged , e.g. as generated by whatshap. The respective suffixes ".genome1", ".genome2", ".unassigned", ".alelle_flagged" are required to be followed by the bam extention ".sorted.bam". This mode is mutually exclusive with "deepTools_qc". Only the allelic version of deepTools qc will be run, by default. Allelic version of featureCounts will be run by default. If sample sheet is provided, allelic DESeq2- or allelic Salmon-based differential gene expression analysis will be run.
"alignment-free"¶
In this mode, the pipeline uses salmon to perform transcript-level expression quantification. This mode performs both transcript-level differential expression (using Sleuth), and gene-level differential expression (using wasabi, followed by DESeq2).
Note
The salmon index is recreated each time in alignment-free mode. This is done to facilitate changing how the GTF file is filtered, which necessitates reindexing.
"deepTools_qc"¶
The pipeline provides multiple quality controls through deepTools, which can be triggered using the deepTools_qc mode. It's a very useful add-on with any of the other modes.
Note
Since most deeptools functions require an aligned (BAM) file, the deepTools_qc mode will additionally perform the alignment of the fastq files. However this would not interfere with operations of the other modes.
"threePrimeSeq"¶
threePrimeSeq uses a pipeline developed by the Hilgers lab to annotate and count clusters of reads mapping to three prime ends of genes using poly(T)VN-primed 3' sequencing kits such as Lexogen's 3' mRNA-seq kit. In this mode, fastp is used to pretrim with preset parameters, followed by STAR mapping.
First, a blacklist of possible internal priming sites is generated for the given organism. Next, the mapped regions are filtered according to this blacklist and associated with the nearest gene within a certain window. For all samples within the run, a database of PAS sites is generated and read counts aggregated for each particular site. These are then summarized on a metagene level and output to a counts.tsv file for further downstream analysis.
The output for this mode will be stored in the three_prime_seq/ subfolder.
Note
The --three-prime-seq option must be invoked (which will also set mode to threePrimeSeq) as this will set fastp and STAR with the appropriate parameters.
Understanding the outputs¶
Assuming the pipline was run with --mode alignment-free,alignment,deepTools_qc:
├── Annotation
├── bamCoverage
├── cluster_logs
├── deepTools_qc
│ ├── bamPEFragmentSize
│ ├── estimateReadFiltering
│ ├── logs
│ ├── multiBigwigSummary
│ ├── plotCorrelation
│ ├── plotEnrichment
│ └── plotPCA
├── DESeq2_Salmon_sampleSheet
├── DESeq2_sampleSheet
├── FASTQ
├── featureCounts
├── multiQC
├── QC_report
├── mRNA-seq.cluster_config.yaml
├── mRNA-seq.config.yaml
├── mRNA-seq_organism.yaml
├── mRNA-seq_pipeline.pdf
├── mRNA-seq_run-1.log
├── Salmon
├── sleuth_Salmon_sampleSheet
└── STAR
Note
The _sampleSheet suffix for the DESeq2_sampleSheet and sleuth_Salmon_sampleSheet is drawn from the name of the sample sheet you use. So if you instead named the sample sheet mySampleSheet.txt then the folders would be named DESeq2_mySampleSheet and sleuth_Salmon_mySampleSheet. This facilitates using multiple sample sheets.
Apart from the common module outputs (see Running snakePipes), the workflow would produce the following folders:
Annotation: This folder would contain the GTF and BED files used for analysis. In case the file has been filtered using the
--filterGTGTFFoption (see Configuration file), this would contain the filtered files.STAR/HISAT2: (not produced in mode alignment-free) This would contain the output of RNA-alignment by STAR or HISAT2 (indexed BAM files).
featureCounts: (not produced in mode alignment-free) This would contain the gene-level counts (output of featureCounts), on the filtered GTF files, that can be used for differential expression analysis.
bamCoverage: (not produced in mode alignment-free) This would contain the bigWigs produced by deepTools bamCoverage . Files with suffix
.coverage.bware raw coverage files, while the files with suffixRPKM.bware RPKM-normalized coverage files.deepTools_QC: (produced in the mode deepTools_QC) This contains the quality checks specific for mRNA-seq, performed via deepTools. The output folders are names after various deepTools functions and the outputs are explained under deepTools documentation. In short, they show the insert size distribution(bamPEFragmentSize), mapping statistics (estimateReadFiltering), sample-to-sample correlations and PCA (multiBigwigSummary, plotCorrelation, plotPCA), and read enrichment on various genic features (plotEnrichment)
DESeq2_[sampleSheet]/DESeq2_Salmon_[sampleSheet]: (produced in the modes alignment or alignment-free, only if a sample-sheet is provided.) The folder contains the HTML result report DESeq2_report.html, the annotated output file from DESeq2 (DEseq_basic_DEresults.tsv) and normalized counts for all samples, produced via DEseq2 (DEseq_basic_counts_DESeq2.normalized.tsv) as well as an Rdata file (DEseq_basic_DESeq.Rdata) with the R objects
dds <- DESeq2::DESeq(dds)andddr <- DDESeq2::results(dds,alpha = fdr). DESeq2_[sampleSheet] uses gene counts fromfeatureCounts/counts.tsv, whereas DESeq2_Salmon_[sampleSheet] uses transcript counts fromSalmon/counts.tsvthat are merged via tximport in R. Sample name to plotting shape mapping on the PCA plot is limited to 36 samples and skipped otherwise.Salmon: (produced in mode alignment-free) This folder contains transcript-level (
counts.tsv)and gene-level (counts.genes.tsv) counts estimated by the tool Salmon .sleuth_Salmon_[sampleSheet] (produced in mode alignment-free, only if a sample-sheet is provided) This folder contains a transcript-level differential expression output produced using the tool Sleuth .
Command line options¶
MPI-IE workflow for RNA mapping and analysis
- usage example:
RNA-seq -i input-dir -o output-dir mm10
usage: mRNA-seq -i INDIR -o OUTDIR [-h] [-v] [--ext EXT] [--reads READS READS]
[-c CONFIGFILE] [--clusterConfigFile CLUSTERCONFIGFILE]
[-j INT] [--local] [--keepTemp]
[--snakemakeOptions SNAKEMAKEOPTIONS] [--DAG] [--version]
[--emailAddress EMAILADDRESS] [--smtpServer SMTPSERVER]
[--smtpPort SMTPPORT] [--onlySSL] [--emailSender EMAILSENDER]
[--smtpUsername SMTPUSERNAME] [--smtpPassword SMTPPASSWORD]
[--VCFfile VCFFILE] [--strains STRAINS] [--SNPfile SNPFILE]
[--NMaskedIndex NMASKEDINDEX] [-m MODE] [--downsample INT]
[--trim] [--trimmer {cutadapt,trimgalore,fastp}]
[--trimmerOptions TRIMMEROPTIONS] [--fastqc] [--bcExtract]
[--bcPattern BCPATTERN] [--UMIDedup]
[--UMIDedupSep UMIDEDUPSEP] [--UMIDedupOpts UMIDEDUPOPTS]
[--bwBinSize BWBINSIZE] [--plotFormat STR]
[--libraryType LIBRARYTYPE] [--aligner {STAR,HISAT2}]
[--alignerOptions ALIGNEROPTIONS]
[--featureCountsOptions FEATURECOUNTSOPTIONS]
[--filterGTF FILTERGTF] [--sampleSheet SAMPLESHEET]
[--formula FORMULA] [--dnaContam] [--fromBAM]
[--bamExt BAMEXT] [--singleEnd] [--rMats] [--FDR FDR]
GENOME
Positional Arguments¶
- GENOME
Genome acronym of the target organism. Either a yaml file or one of: hg38, mm39_ens106, GRCh38_gencode40, dm6, GRCz10, SchizoSPombe_ASM294v2, mm10_gencodeM19, GRCz11
Required Arguments¶
- -i, --input-dir
input directory containing the FASTQ files, either paired-end OR single-end data
- -o, --output-dir
output directory
General Arguments¶
- -v, --verbose
verbose output (default: 'False')
- --ext
Suffix used by input fastq files (default: '".fastq.gz"').
- --reads
Suffix used to denote reads 1 and 2 for paired-end data. This should typically be either '_1' '_2' or '_R1' '_R2' (default: '['_R1', '_R2']). Note that you should NOT separate the values by a comma (use a space) or enclose them in brackets.
- -c, --configFile
configuration file: config.yaml (default: 'None')
- --clusterConfigFile
configuration file for cluster usage. In absence, the default options specified in defaults.yaml and workflows/[workflow]/cluster.yaml would be selected (default: 'None')
- -j, --jobs
maximum number of concurrently submitted Slurm jobs / cores if workflow is run locally (default: '5')
- --local
run workflow locally; default: jobs are submitted to Slurm queue (default: 'False')
- --keepTemp
Prevent snakemake from removing files marked as being temporary (typically intermediate files that are rarely needed by end users). This is mostly useful for debugging problems.
- --snakemakeOptions
Snakemake options to be passed directly to snakemake, e.g. use --snakemakeOptions='--dryrun --rerun-incomplete --unlock --forceall'. WARNING! ONLY EXPERT USERS SHOULD CHANGE THIS! THE DEFAULT VALUE WILL BE APPENDED RATHER THAN OVERWRITTEN! (default: '['--use-conda']')
- --DAG
If specified, a file ending in _pipeline.pdf is produced in the output directory that shows the rules used and their relationship to each other.
- --version
show program's version number and exit
Email Arguments¶
- --emailAddress
If specified, send an email upon completion to the given email address
- --smtpServer
If specified, the email server to use.
- --smtpPort
The port on the SMTP server to connect to. A value of 0 specifies the default port.
- --onlySSL
The SMTP server requires an SSL connection from the beginning.
- --emailSender
The address of the email sender. If not specified, it will be the address indicated by --emailAddress
- --smtpUsername
If your SMTP server requires authentication, this is the username to use.
- --smtpPassword
If your SMTP server requires authentication, this is the password to use.
Allele-specific mapping arguments¶
- --VCFfile
VCF file to create N-masked genomes (default: 'None')
- --strains
Name or ID of SNP strains separated by comma (default: 'None')
- --SNPfile
File containing SNP locations (default: 'None')
- --NMaskedIndex
N-masked index of the reference genome (default: 'None'). Note that this should point to a file (i.e. 'Genome' for STAR indices, genome.1.bt2 for bowtie2 indices).
Options¶
- -m, --mode
workflow running modes (available: 'alignment-free, alignment, allelic-mapping, allelic-counting, deepTools_qc, three-prime-seq') (default: '"alignment,deepTools_qc"')
- --downsample
Downsample the given number of reads randomly from of each FASTQ file (default: 'False')
- --trim
Activate fastq read trimming. If activated, Illumina adaptors are trimmed by default. Additional parameters can be specified under --trimmerOptions. (default: 'False')
- --trimmer
Possible choices: cutadapt, trimgalore, fastp
Trimming program to use: Cutadapt, TrimGalore, or fastp. Note that if you change this you may need to change --trimmerOptions to match! (default: '"cutadapt"')
- --trimmerOptions
Additional option string for trimming program of choice. (default: 'None')
- --fastqc
Run FastQC read quality control (default: 'False')
- --bcExtract
To extract umi barcode from fastq file via UMI-tools and add it to the read name (default: 'False')
- --bcPattern
The pattern to be considered for the barcode. 'N' = UMI position (required) 'C' = barcode position (optional) (default: '"NNNNCCCCCCCCC"')
- --UMIDedup
Deduplicate bam file based on UMIs via umi_tools dedup that are present in the read name. (default: 'False')
- --UMIDedupSep
umi separation character that will be passed to umi_tools.(default: '"_"')
- --UMIDedupOpts
Additional options that will be passed to umi_tools.(default: '')
- --bwBinSize
Bin size of output files in bigWig format (default: '25')
- --plotFormat
Possible choices: png, pdf, None
Format of the output plots from deepTools. Select 'none' for no plots (default: '"png"')
- --libraryType
user provided library type strand specificity. featureCounts style: 0, 1, 2 (Illumina TruSeq); default: '2')
- --aligner
Possible choices: STAR, HISAT2
Program used for mapping: STAR or HISAT2 (default: '"STAR"'). If you change this, please change --alignerOptions to match.
- --alignerOptions
STAR or hisat2 option string, e.g.: '--twopassMode Basic' (default: 'None')
- --featureCountsOptions
featureCounts option string. The options '-p -B' are always used for paired-end data (default: '"-C -Q 10 --primary"')
- --filterGTF
filter annotation GTF by grep for use with Salmon, e.g. use --filterGTF='-v pseudogene'; default: 'None')
- --sampleSheet
Information on samples (required for DE analysis); see 'https://github.com/maxplanck-ie/snakepipes/tree/master/docs/content/sampleSheet.example.tsv' for example. The column names in the tsv files are 'name' and 'condition'. The first entry defines which group of samples are control. This way, the order of comparison and likewise the sign of values can be changed. The DE analysis might fail if your sample names begin with a number. So watch out for that! (default: 'None')
- --formula
Design formula to use in linear model fit (default: 'None')
- --dnaContam
Returns a plot which presents the proportion of the intergenic reads (default: 'False')
- --fromBAM
Input folder with bam files. If provided, the analysis will start from this point. If bam files contain single ends, please specify --singleEnd additionally.
- --bamExt
Extention of provided bam files, will be subtracted from basenames to obtain sample names. (default: '".bam"')
- --singleEnd
input data is single-end, not paired-end. This is only used if --fromBAM is specified.
- --rMats
Run differential splicing analysis using rMats-turbo. Note that this flag requires --sampleSheet to be specified.
- --FDR
FDR threshold to apply for filtering DE genes(default: '0.05')