preprocessing
What it does
The preprocessing pipeline handles a few tasks that are commonly done by some, but not all, sequencing providers:
* Merging samples across lanes (or technical replicates)
* Removal of apparent optical duplicates
* Reformatting fastq files to extract UMIs
* Running FastQC
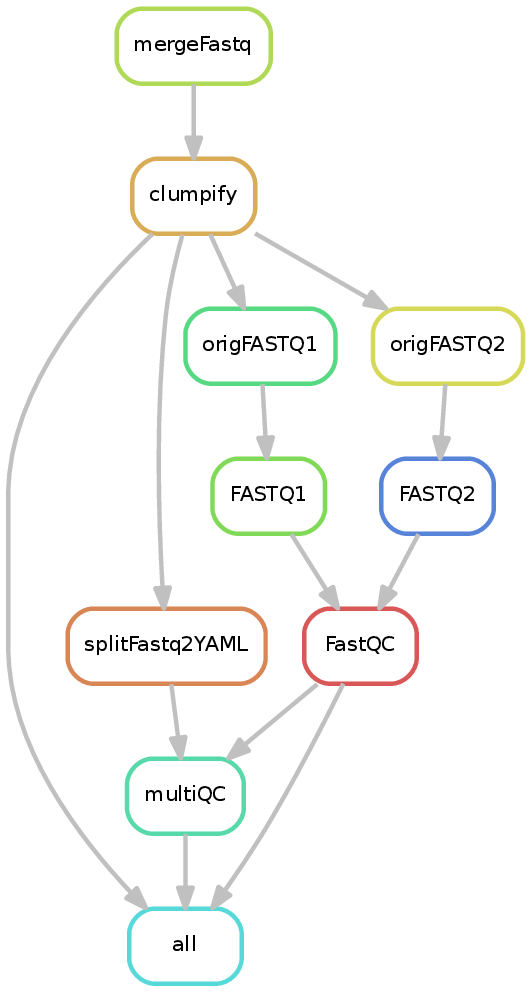
Input requirements
The minimal requirement is a directory of fastq files. If files should be merged (e.g., the sequencing provider did not merge samples across lanes) then a sample sheet should also be provided of the following form:
sample1_S1_L001_R1_001.fastq.gz _R1 sample1
sample1_S1_L001_R2_001.fastq.gz _R2 sample1
sample1_S1_L002_R1_001.fastq.gz _R1 sample1
sample1_S1_L002_R2_001.fastq.gz _R2 sample1
sample1_S1_L003_R1_001.fastq.gz _R1 sample1
sample1_S1_L003_R2_001.fastq.gz _R2 sample1
The first column contains file names, the second the associated read 1/2 designator (this should match the --reads option), and finally the desired sample name.
Care should be given when setting --optDedupDist, as values of 0 (the default) disable removal of optical duplicates. The appropriate value to use is sequencer-dependent.
Understanding the outputs
The preprocessing pipeline can generate the following files and directories (depending on the options given):
.
├── deduplicatedFASTQ
│ ├── sample1.metrics
│ ├── sample1_R1.fastq.gz
│ ├── sample1_R1_optical_duplicates.fastq.gz
│ ├── sample1_R2.fastq.gz
│ ├── sample1_R2_optical_duplicates.fastq.gz
│ └── optical_dedup_mqc.json
├── FASTQ
├── FastQC
├── mergedFASTQ
├── multiQC
└── originalFASTQ
As shown above, the pipeline produces the following directories:
mergedFASTQ : If a sample sheet is given, this file contains the merged fastq files.
deduplicatedFASTQ: The results of optical duplicate removal (or symlinks to
mergedFASTQ). The "_optical_duplicates" files contain the reads marked by clumpify as being likely optical duplicates. The associated ".metrics" file contains two values: number of optical duplicates and then the total reads. Theoptical_dedup_mqc.jsonfile merges the various sample metrics files for downstream use by MultiQC.originalFASTQ : This folder exists from compatibility with other pipelines and will contain either symlinks to the original fastq files or, if a sample sheet is specified, those in
deduplicatedFASTQ.FASTQ: Fastq files produced by UMI processing (or symlinks to
originalFASTQ).FastQC: If the
--fastqcparameter was given, the output of FastQC.multiQC: If either FastQC was run or optical duplicates were removed, an interactive web report will be created using MultiQC.
Command line options
MPI-IE workflow for preprocessing
- usage example:
preprocessing -i input-dir -o output-dir --optDedupDist 2500 --clumpifyOptions
usage: Preprocessing -i INDIR -o OUTDIR [-h] [-v] [--ext EXT]
[--reads READS READS] [-c CONFIGFILE] [--keepTemp]
[--snakemakeOptions SNAKEMAKEOPTIONS] [--DAG] [--version]
[--emailAddress EMAILADDRESS] [--smtpServer SMTPSERVER]
[--smtpPort SMTPPORT] [--onlySSL]
[--emailSender EMAILSENDER] [--smtpUsername SMTPUSERNAME]
[--smtpPassword SMTPPASSWORD] [--fastqc] [--bcExtract]
[--bcPattern BCPATTERN] [--optDedupDist OPTDEDUPDIST]
[--clumpifyOptions CLUMPIFYOPTIONS]
[--clumpifyMemory CLUMPIFYMEMORY]
[--sampleSheet SAMPLESHEET | --automaticIlluminaMerging]
Required Arguments
- -i, --input-dir
input directory containing the FASTQ files, either paired-end OR single-end data
- -o, --output-dir
output directory
General Arguments
- -v, --verbose
verbose output (default: 'False')
- --ext
Suffix used by input fastq files (default: ''.fastq.gz'').
- --reads
Suffix used to denote reads 1 and 2 for paired-end data. This should typically be either '_1' '_2' or '_R1' '_R2' (default: '['_R1', '_R2']). Note that you should NOT separate the values by a comma (use a space) or enclose them in brackets.
- -c, --configFile
configuration file: config.yaml (default: 'None')
- --keepTemp
Prevent snakemake from removing files marked as being temporary (typically intermediate files that are rarely needed by end users). This is mostly useful for debugging problems.
- --snakemakeOptions
Snakemake options to be passed directly to snakemake, e.g. use --snakemakeOptions='--dryrun --rerun-incomplete --unlock --forceall'. WARNING! ONLY EXPERT USERS SHOULD CHANGE THIS! THE DEFAULT VALUE WILL BE APPENDED RATHER THAN OVERWRITTEN! (default: '['--use-conda']')
- --DAG
If specified, a file ending in _pipeline.pdf is produced in the output directory that shows the rules used and their relationship to each other.
- --version
show program's version number and exit
Email Arguments
- --emailAddress
If specified, send an email upon completion to the given email address
- --smtpServer
If specified, the email server to use.
- --smtpPort
The port on the SMTP server to connect to. A value of 0 specifies the default port.
- --onlySSL
The SMTP server requires an SSL connection from the beginning.
- --emailSender
The address of the email sender. If not specified, it will be the address indicated by --emailAddress
- --smtpUsername
If your SMTP server requires authentication, this is the username to use.
- --smtpPassword
If your SMTP server requires authentication, this is the password to use.
Options
- --fastqc
Run FastQC read quality control (default: 'False')
- --bcExtract
To extract umi barcode from fastq file via UMI-tools and add it to the read name (default: 'False')
- --bcPattern
The pattern to be considered for the barcode. 'N' = UMI position (required) 'C' = barcode position (optional) (default: '''')
- --optDedupDist
The maximum distance between clusters to mark one as an optical duplicate of the other. Setting this to 0 will disable optical deduplication, which is only needed on patterned flow cells (NextSeq, NovaSeq, HiSeq 3000/4000, etc.). Common values are: 2500 (HiSeq 3000/4000), 40 (NextSeq) or 12000 (NovaSeq). (default: '0')
- --clumpifyOptions
Options passed to clumpify, which should generally NOT be changed. The exception to this is with NextSeq runs, where 'spany=t adjacent=t' should be ADDED. (default: ''dupesubs=0 qin=33 markduplicates=t optical=t')
- --clumpifyMemory
This controls how much memory clumpify is instructed to use, in GB. This may need to be increased if samples are particularly large or there are MANY optical duplicates. This is passed to clumpify (e.g., as -Xmx30G). You may additionally need to instruct your scheduler about the per-core memory usage (e.g., in cluster.yaml). (default: ''30G'')
- --sampleSheet
Information on samples (required for merging across lanes); see 'docs/content/preprocessing_sampleSheet.example.tsv' for an example. The first set of columns should hold the current sample names (excluding things like _R1.fastq.gz or _1.fastq.gz) while the second holds the name that the final sample should have (again, excluding things like _R1.fastq.gz or _1.fastq.gz). (default: 'None')
- --automaticIlluminaMerging
Instead of manually specifying a sample sheet that can be used to merge samples, this option assumes that fastq files in the input directory follow the default output of bcl2fastq from Illumina and should be merged across lanes. An example of this would be that files named Sample1_S1_L001_R1_001.fastq.gz and Sample1_S1_L002_R1_001.fastq.gz would be merged to Sample1_R1.fastq.gz. This option CAN NOT be combined with --sampleSheet. It is assumed that standard R1/R2 read designators are being used and that all files end in .fastq.gz