scRNA-seq¶
What it does¶
The scRNA-seq pipeline is intended to process UMI-based data, expecting the cell barcode and umi in Read1, and the cDNA sequence in Read2. The workflow has predefined settings for CelSeq2 and 10x data, but can be extended to custom protocols.
There are currently two analysis modes available: - "STARsolo" which uses STAR solo for mapping and quantitation. - "Alevin" based on Salmon for generating the count matrix.
Note
Mode "Gruen" has been deprecated.
The general procedure for mode "STARsolo" involves:
moving cell barcodes and UMIs from read 1 into the CB and UMI tags of read 2 during mapping (STARsolo),
quantification of genic read counts at the single cell level (STARsolo),
quantification of reads supporting spliced and unspliced transcripts in each cell (velocyto) - unless this has been disabled with --skipVelocyto
generation of seurat objects for genic counts.
UMIs in the read headers are used to avoid counting PCR duplicates. A number of bigWig and QC plots (e.g., from plotEnrichment) are generated as well.
Mode "Alevin" involves:
Generation of a salmon index used for mapping.
Mapping and generation of a readcount matrix.
Estimation of uncertainty of gene counts using bootstrap method implemented in Salmon Alevin.
General QC of the Alevin run using the AlevinQC R package.
Quantification of "spliced" and "unspliced" read counts in each cell with Alevin - unless this has been disabled with --skipVelocyto . This analysis is derived from the code underlying Soneson et al. 2020, bioRxiv https://doi.org/10.1101/2020.03.13.990069.
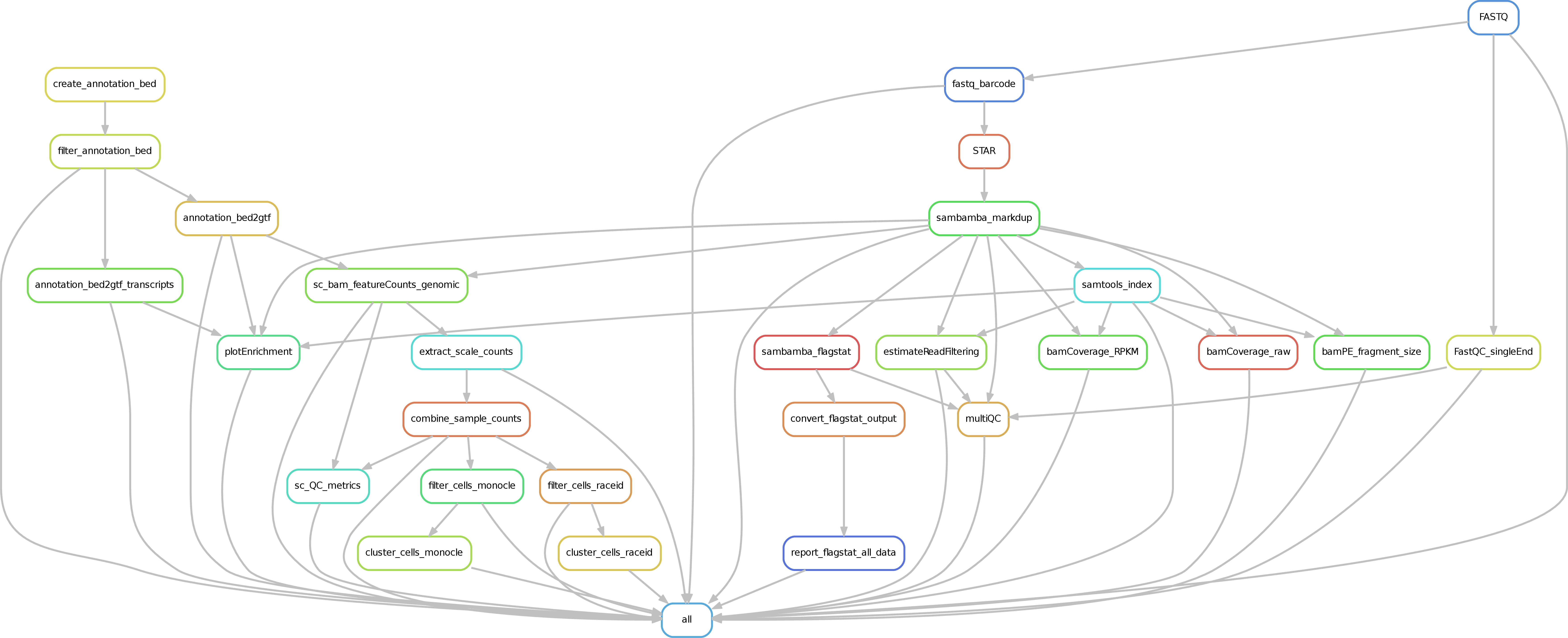
Mode STARsolo¶
With current settings, this mode should work with any UMI-based protocol that stores UMI and CB in read 1, each in one chunk. The mode comes with four presets that can be passed to the ' --myKit ' argument: CellSeq192, CellSeq384, 10xV2, 10xV3. Choosing a preset will select a corresponding barcode whitelist file as well as cell barcode and umi length and positions to be used. Choosing the Custom preset allows the user to run the workflow providing own barcode whitelist and CB/UMI positions and lengths. CellSeq384 is the current default preset.
In this mode, STARsolo is used to map, UMI-deduplicate and count reads. Importantly, read 1 is expected to carry the UMI and the cell barcode, while read 2 is expected to carry the cDNA sequence. Default positions of UMI and CB in read 1 are specified, as well as their respective lengths. If your setup is different from the available presets, change it via the --STARsoloCoords commandline argument or in the defaults.yaml dictionary, in addition to providing --myKit Custom argument.
In the STARsolo folder, bam files are stored, along with 10x-format count matrices and log files summarizing barcode detection and UMI-deduplication. Bam files have the UB and CB tags set.
Deeptools QC is run on these bam files.
Before running velocyto, bam files from STARsolo are filtered to remove unmapped reads as well as reads with an empty CB tag and then cell-sorted by the CB tag. In the VelocytoCounts folder, loom files with counts of spliced, unspliced and ambiguous reads are stored. A merged loom file containing counts for all samples together can be found in the VelocytoCounts_merged folder. As Velocyto tends to consume a lot of memory and result in long runtimes with cell numbers in ~10^5, it can be disabled with --skipVelocyto.
Input requirements¶
The primary input requirement is a directory of paired-end fastq files. For the STAR solo mode, a barcode whitelist is required, as well as specification of UMI and CB positions and length, if different from default or available presets.
Barcode whitelist¶
Required for the STARsolo mode. The expected format is a one-column txt file with barcodes the user wishes to retain. Default is a whitelist file for CellSeq2 384 barcodes, provided with the pipeline. If 'myKit' is changed to another available preset, the corresponding barcode whitelist provided with the pipeline will be used.
Configuration file¶
The default configuration file is listed below and can be found in snakePipes/workflows/scRNAseq/defaults.yaml:
pipeline: scrna-seq
outdir:
configFile:
clusterConfigFile:
local: False
maxJobs: 5
## directory with fastq files
indir:
## preconfigured target genomes (mm9,mm10,dm3,...) , see /path/to/snakemake_workflows/shared/organisms/
## Value can be also path to your own genome config file!
genome:
## FASTQ file extension (default: ".fastq.gz")
ext: '.fastq.gz'
## paired-end read name extension (default: ["_R1", "_R2"])
reads: ["_R1","_R2"]
##Analysis mode
mode: STARsolo
## Number of reads to downsample from each FASTQ file
downsample:
## Options for trimming
trim: False
trimmer: cutadapt
trimmerOptions: -a A{'30'}
## --twopassMode Basic is not compatible with --outStd in all STAR versions
alignerOptions: ""
## further options
filterGTF: "-v -P 'decay|pseudogene' "
cellBarcodeFile:
cellBarcodePattern: "NNNNNNXXXXXX"
splitLib: False
cellNames:
##mode STARsolo options
myKit: CellSeq384
BCwhiteList:
STARsoloCoords: ["1","7","8","7"]
skipVelocyto: False
##mode Alevin options
alevinLibraryType: "ISR"
prepProtocol: "chromiumV3"
expectCells:
readLengthFrx: 0.2
#generic options
libraryType: 1
bwBinSize: 10
verbose: False
plotFormat: pdf
dnaContam: False
## Parameters for th statistical analysis
cellFilterMetric: gene_universe
#Option to skip RaceID to save time
skipRaceID: False
#umi_tools options:
UMIBarcode: False
bcPattern: NNNNCCCCCCCCC #default: 4 base umi barcode, 9 base cell barcode (eg. RELACS barcode)
UMIDedup: False
UMIDedupSep: "_"
UMIDedupOpts: --pairedUMIDedupOpts: --paired
Pseudogene filter¶
As default, transcripts or genes that contain that are related to biotypes like 'pseudogene' or 'decay' are filtered out before tag counting (see
--filterGTF default).
Here we assume you provide eg. a gencode or ensemble annotation file (via genes_gtf in the organism configuration yaml) that contains this information.
Library Type¶
The CEL-seq2 protocol produces reads where read 2 maps in sense direction (libraryType: 1).
Fraction of read length required to overlap the intron¶
In mode Alevin, the fraction of read length required to overlap the intron in order to be counted as "unspliced" is set to 0.2 (i.e. 20%) by default. This corresponds to 10nt in a 50nt-long read, or to 20nt in a 100nt-long read. The user is encouraged to modify this value as deemed appropriate via the --readLengthFrx commandline argument.
In practice, this variable affects the length of the exon sequence flank added to the intron sequence to generate reference sequences for Salmon Alevin. Exon sequence flank length is set to one minus 'readLengthFrx' of read length.
Output structure¶
The following will be produced in the output directory when the workflow is run in mode STARsolo:
analysis/
├── scRNAseq_run-1.log
├── multiQC
├── deepTools_qc
├── cluster_logs
├── bamCoverage
├── Sambamba
├── filtered_bam
├── STARsolo
├── Seurat
├── Annotation
├── FastQC
├── originalFASTQ
├── scRNAseq_tools.txt
├── scRNAseq.cluster_config.yaml
├── scRNAseq.config.yaml
└── scRNAseq_organism.yaml
- The **VelocytoCounts** directory contains loom files in sample subdirectories.
- The **VelocytoCounts_merged** directory containes one loom file with all samples merged.
- The **STARsolo** directory contains bam files and 10X-format cell count matrices produced by STARsolo.
- The **Annotation** directory contains a filtered version of your original GTF file, with pseudogenes removed by default.
- The **bamCoverage** directory contains a bigwig track for each sample (not per cell!). This can be used eg. in IGV to check where your reads map in general.
- The **deeptools_qc** directory contains additional QC reports and plots. The ``FASTQC`` directory can be used to verify eg. the barcode layout of read 1.
The following output structure will be produced when running in Alevin mode:
├── Alevin
├── AlevinForVelocity
├── Annotation
├── cluster_logs
├── FastQC
├── multiQC
├── originalFASTQ
├── Salmon
├── scRNAseq.cluster_config.yaml
├── scRNAseq.config.yaml
├── scRNAseq_organism.yaml
├── scRNAseq_pipeline.pdf
├── scRNAseq_run-1.log
├── scRNAseq_tools.txt
└── SingleCellExperiment
- The **Salmon** directory contains the generated genome index.
- The **Alevin** directory contains the matrix files (both bootstrapped and raw) per sample in subdirectories.
- The **multiQC** directory contains an additional alevinQC html file generated per sample.
- The **AlevinForVelocity** directory contains the matrix files with "spliced" and "unspliced" reads per cell in subdirectories.
- The **SingleCellExperiment** directory contains the RDS files with "SingleCellExperiment" class R objects, storing spliced/unspliced counts per cell in corresponding assays.
Understanding the outputs: mode STARsolo¶
Main result: output folders with 10x-format count matrices can be found in sample subfolders under
STARsolo. The ouput consists of three files: barcodes.tsv, features.tsv, matrix.mtx. Their gzipped versions are stored in the same folder. Seurat objects from merged samples are available in theSeuratfolder.Corresponding annotation files are:
Annotation/genes.filtered.bedandAnnotation/genes.filtered.gtf, respectively.The folders
QC_report,FASTQC,deeptools_qcandmultiQCcontain various QC tables and plots.STARsolo directory contain the output from genomic alignments.
Understanding the outputs: mode Alevin¶
Main result: output folders containing the raw and boostrapped count matrices are found under the sample subfolders under
Alevin. The sample specific Alevin folders contain the matrices, as well as column data (barcodes) and row data (genes). Alevin spliced/unspliced counts for RNA velocity are stored as alevin matrices in the sample subfolders underAlevinForVelocityand as "SingleCellExperiment" class R objects underSingleCellExperiment.Corresponding annotation files are:
Annotation/genes.filtered.bedandAnnotation/genes.filtered.gtf, respectively.The QC plots (both from multiQC and AlevinQC) are available in the
multiQCfolder.
Command line options¶
MPI-IE workflow for scRNA-seq (CEL-Seq2 and related protocols)
- usage example:
scRNAseq -i input-dir -o output-dir mm10
usage: scRNAseq -i INDIR -o OUTDIR [-h] [-v] [--ext EXT] [--reads READS READS]
[-c CONFIGFILE] [--clusterConfigFile CLUSTERCONFIGFILE]
[-j INT] [--local] [--keepTemp]
[--snakemakeOptions SNAKEMAKEOPTIONS] [--DAG] [--version]
[--emailAddress EMAILADDRESS] [--smtpServer SMTPSERVER]
[--smtpPort SMTPPORT] [--onlySSL] [--emailSender EMAILSENDER]
[--smtpUsername SMTPUSERNAME] [--smtpPassword SMTPPASSWORD]
[--mode STR] [--downsample INT] [--filterGTF FILTERGTF]
[--BCwhiteList STR] [--STARsoloCoords STARSOLOCOORDS]
[--bwBinSize BWBINSIZE] [--plotFormat STR] [--myKit STR]
[--skipVelocyto]
[--prepProtocol {dropseq,chromiumV3,chromium,gemcode,citeseq,celseq,celseq2,quartzseq2}]
[--alevinLibraryType {ISR,ISF,MSF,MSR,OSR,OSF}]
[--expectCells EXPECTCELLS]
GENOME
Positional Arguments¶
- GENOME
Genome acronym of the target organism. Either a yaml file or one of: hg38, mm39_ens106, GRCh38_gencode40, dm6, GRCz10, SchizoSPombe_ASM294v2, mm10_gencodeM19, GRCz11
Required Arguments¶
- -i, --input-dir
input directory containing the FASTQ files, either paired-end OR single-end data
- -o, --output-dir
output directory
General Arguments¶
- -v, --verbose
verbose output (default: 'False')
- --ext
Suffix used by input fastq files (default: '".fastq.gz"').
- --reads
Suffix used to denote reads 1 and 2 for paired-end data. This should typically be either '_1' '_2' or '_R1' '_R2' (default: '['_R1', '_R2']). Note that you should NOT separate the values by a comma (use a space) or enclose them in brackets.
- -c, --configFile
configuration file: config.yaml (default: 'None')
- --clusterConfigFile
configuration file for cluster usage. In absence, the default options specified in defaults.yaml and workflows/[workflow]/cluster.yaml would be selected (default: 'None')
- -j, --jobs
maximum number of concurrently submitted Slurm jobs / cores if workflow is run locally (default: '5')
- --local
run workflow locally; default: jobs are submitted to Slurm queue (default: 'False')
- --keepTemp
Prevent snakemake from removing files marked as being temporary (typically intermediate files that are rarely needed by end users). This is mostly useful for debugging problems.
- --snakemakeOptions
Snakemake options to be passed directly to snakemake, e.g. use --snakemakeOptions='--dryrun --rerun-incomplete --unlock --forceall'. WARNING! ONLY EXPERT USERS SHOULD CHANGE THIS! THE DEFAULT VALUE WILL BE APPENDED RATHER THAN OVERWRITTEN! (default: '['--use-conda']')
- --DAG
If specified, a file ending in _pipeline.pdf is produced in the output directory that shows the rules used and their relationship to each other.
- --version
show program's version number and exit
Email Arguments¶
- --emailAddress
If specified, send an email upon completion to the given email address
- --smtpServer
If specified, the email server to use.
- --smtpPort
The port on the SMTP server to connect to. A value of 0 specifies the default port.
- --onlySSL
The SMTP server requires an SSL connection from the beginning.
- --emailSender
The address of the email sender. If not specified, it will be the address indicated by --emailAddress
- --smtpUsername
If your SMTP server requires authentication, this is the username to use.
- --smtpPassword
If your SMTP server requires authentication, this is the password to use.
Options¶
- --mode
Possible choices: STARsolo, Alevin
Analysis mode. Possible settings are 'Gruen, STARsolo and Alevin' Default: "STARsolo"
- --downsample
Downsample the given number of reads randomly from of each FASTQ file
- --filterGTF
filter annotation GTF by grep for feature counting, e.g. use --filterGTF='-v pseudogene'; (default: '"-v -P 'decay|pseudogene' "')
- --BCwhiteList
Path to a one-column txt file with barcode whitelist. Required for the STARsolo mode,optional for Alevin mode. (default: 'None')
- --STARsoloCoords
Comma-separated list of values: UMI start position, UMI length, CB start position, CB length. Required for the STARsolo mode (default: '['1', '7', '8', '7']')
- --bwBinSize
Bin size of output files in bigWig format (default: '10')
- --plotFormat
Possible choices: png, pdf, None
Format of the output plots from deeptools. Select 'none' for no plot (default: '"png"')
- --myKit
Possible choices: 10Xv2, 10Xv3, CellSeq192, CellSeq384, Custom
Library preparation kit and version to use preset barcode whitelist and CB/UMI positions for (default: '"CellSeq384"')
- --skipVelocyto
Skip bam filtering and generating RNA velocity counts by velocyto to save time and memory usage. (default: 'False')
- --prepProtocol
Possible choices: dropseq, chromiumV3, chromium, gemcode, citeseq, celseq, celseq2, quartzseq2
Alevin mode. Specify the library prep method. (default: 'None')
- --alevinLibraryType
Possible choices: ISR, ISF, MSF, MSR, OSR, OSF
Alevin mode. Library orientation type. (default: '"ISR"')
- --expectCells
Alevin mode. Optional to fill in if you know how many cells are expected. (default: 'None')