ATAC-seq¶
What it does¶
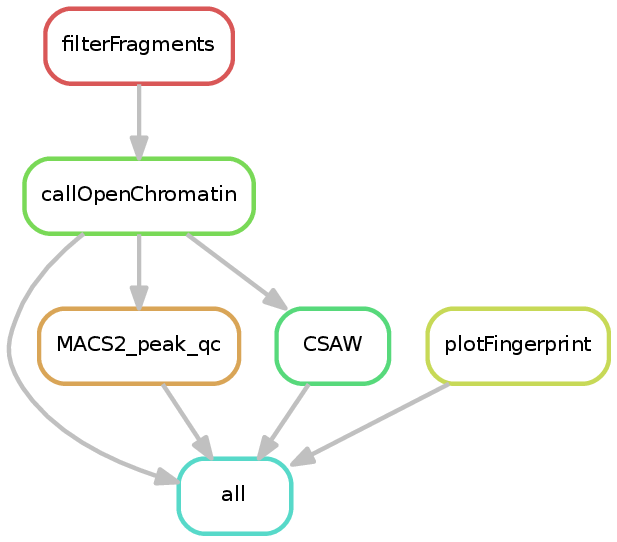
The ATAC-seq pipeline takes one or more BAM files and attempts to find accessible regions. If multiple samples and a sample sheet are provided, then CSAW is additionally used to find differentially accessible regions. Prior to finding open/accessible regions, the BAM files are filtered to include only properly paired reads with appropriate fragment sizes (<150 bases by default). These filtered fragments are then used for the remainder of the pipeline.
Note
The CSAW step will be skipped if there is no sample_info tsv file (see Running snakePipes).
Input requirements¶
The DNA mapping pipeline generates output that is fully compatible with the ATAC-seq pipeline input requirements!
When running the ATAC-seq pipeline, please specify the output directory of DNA-mapping pipeline as the working directory (-d).
filtered_bam directory contains the input BAM files (either filtered or unfiltered, however you prefer).
sampleSheet.tsv (OPTIONAL) is only needed to test for differential binding.
Differential open chromatin analysis¶
Similar to differential binding analysis with the ChIP-Seq data. We can perform the differential open chromatin analysis, using the --sampleSheet option of the ATAC-seq workflow. This requires a sample sheet, which is identical to that required by the ChIP-seq and RNA-seq workflows (see ChIP-seq for details).
An example is below:
name condition
sample1 eworo
sample2 eworo
SRR7013047 eworo
SRR7013048 OreR
SRR7013049 OreR
SRR7013050 OreR
Note
This sample sheet has the same requirements as the sample sheet in the ChIP-seq workflow, and also uses the same tool (CSAW) with a narrow default window size.
For comparison between two conditions, the name you assign to "condition" is not relevant, but rather the order is. The group mentioned first (in the above case "wild-type") would be used as a "control" and the group mentioned later would be used as "test".
If the user provides additional columns between 'name' and 'condition' in the sample sheet, the variables stored there will be used as blocking factors in the order they appear in the sample sheet. Condition will be the final column and it will be used for any statistical inference.
The differential binding module utilizes the R package CSAW to detect significantly different peaks between two conditions.
The analysis is performed on a union of peaks from all samples mentioned in the sample sheet.
This merged set of regions are provided as an output inside the CSAW_MACS2_sampleSheet folder as the file 'DiffBinding_allregions.bed'.
All differentially bound regions are available in 'CSAW/DiffBinding_significant.bed' .
Two thresholds are applied to produce Filtered.results.bed : FDR (default 0.05 ) as well as absolute log fold change (1). These can be specified either in the defaults.yaml dictionary or via commandline parameters '--FDR' and '--LFC'. Additionally, filtered results are split into up to 3 bed files, representing direction change (UP, DOWN, or MIXED).
Note
In order to include or exclude peaks from selected samples in the union of peaks used in the differential binding analysis, the user may provide an additional column named 'UseRegions' and set it to True or False, accordingly. This column must supersede the 'condition' column in the column order.
Merged regions from filtered results with any direction change are further used to produce deepTools heatmaps, using depth-normalized coverage. For this purpose, the regions are rescaled to 1kb, and extended by 0.2kb on each side.
An html report summarizing the differential binding analysis is produced in the same folder.
Filtered results are also annotated with the distance to the closest gene using bedtools closest and written as '.txt' files to the AnnotatedResults_* folder.
Configuration file¶
There is a configuration file in snakePipes/workflows/ATACseq/defaults.yaml:
## General/Snakemake parameters, only used/set by wrapper or in Snakemake cmdl, but not in Snakefile
pipeline: ATAC-seq
configFile:
clusterConfigFile:
local: false
maxJobs: 5
## workingdir need to be required DNA-mapping output dir, 'outdir' is set to workingdir internally
workingdir:
## preconfigured target genomes (mm9,mm10,dm3,...) , see /path/to/snakemake_workflows/shared/organisms/
## Value can be also path to your own genome config file!
genome:
## The maximum fragment size to retain. This should typically be the size of a nucleosome
maxFragmentSize: 150
minFragmentSize: 0
verbose: false
## which peak caller to use
peakCaller: 'MACS2'
# sampleSheet_DB
sampleSheet:
# windowSize
windowSize: 20
fragmentCountThreshold: 1
#### Flag to control the pipeline entry point
bamExt: '.filtered.bam'
fromBAM:
## Bin size of output files in bigWig format
bwBinSize: 25
pairedEnd: True
plotFormat: png
## Median/mean fragment length, only relevant for single-end data (default: 200)
fragmentLength:
trim:
fastqc:
qval: 0.001
##dummy string to skip filtering annotation
filter_annotation:
##parameters to filter DB regions on
fdr: 0.05
absBestLFC: 1
Useful parameters are maxFragmentSize, minFragmentSize and windowSize, also available from commandline.
windowSize: is the size of windows to test differential binding using CSAW. The default small window size is sufficient for most analysis, since an ATAC-seq peak is sharp.
fragmentCountThreshold: refers to the minimum number of counts a chromosome must have to be included in the MACS2 analysis. It is introduced to avoid errors in the peak calling step and should only be changed if MACS2 fails.
Qval: a value provided to MACS2 that affects the number and width of the resulting peaks.
Understanding the outputs¶
Assuming a sample sheet is used, the following will be added to the working directory:
.
├── CSAW_MACS2_sampleSheet
│ ├── CSAW.log
│ ├── CSAW.session_info.txt
│ ├── DiffBinding_allregions.bed
│ ├── DiffBinding_analysis.Rdata
│ ├── DiffBinding_modelfit.pdf
│ ├── DiffBinding_scores.txt
│ ├── DiffBinding_significant.bed
│ ├── QCplots_first_sample.pdf
│ ├── QCplots_last_sample.pdf
│ └── TMM_normalizedCounts.pdf
├── deepTools_ATAC
│ └── plotFingerprint
│ ├── plotFingerprint.metrics.txt
│ └── plotFingerprint.png
├── Genrich
│ └── group1.narrowPeak
├── HMMRATAC
│ ├── sample1.log
│ ├── sample1.model
│ ├── sample1_peaks.gappedPeak
│ ├── sample1_summits.bed
│ └── sample1_training.bed
├── MACS2
│ ├── sample1.filtered.BAM_control_lambda.bdg
│ ├── sample1.filtered.BAM_peaks.narrowPeak
│ ├── sample1.filtered.BAM_peaks.xls
│ ├── sample1.filtered.BAM_summits.bed
│ ├── sample1.filtered.BAM_treat_pileup.bdg
│ ├── sample1.short.metrics
│ ├── sample2.filtered.BAM_control_lambda.bdg
│ ├── sample2.filtered.BAM_peaks.narrowPeak
│ ├── sample2.filtered.BAM_peaks.xls
│ ├── sample2.filtered.BAM_summits.bed
│ ├── sample2.filtered.BAM_treat_pileup.bdg
│ └── sample2.short.metrics
└── MACS2_QC
├── sample1.filtered.BAM_peaks.qc.txt
└── sample2.filtered.BAM_peaks.qc.txt
Currently the ATAC-seq workflow performs detection of open chromatin regions via MACS2 (or HMMRATAC or Genrich, if specified with --peakCaller), and if a sample sheet is provided, the detection of differential open chromatin sites via CSAW. There are additionally log files in most of the directories. The various outputs are documented in the CSAW and MACS2 documentation.
For more information on the contents of the CSAW_MACS2_sampleSheet folder, see section Differential open chromatin analysis .
MACS2 / HMMRATAC / Genrich: Contains peaks found by the peak caller. The most useful files end in
.narrowPeakor.gappedPeakand are appropriate for visualization in IGV.- MACS2_QC: contains a number of QC metrics that we find useful, namely :
the number of peaks
fraction of reads in peaks (FRiP)
percentage of the genome covered by peaks.
deepTools_ATAC: contains the output of plotFingerPrint, which is a useful QC plot to assess signal enrichment between the ATAC-seq samples.
Note
The _sampleSheet suffix for the CSAW_MACS2_sampleSheet is drawn from the name of the sample sheet you use. So if you instead named the sample sheet mySampleSheet.txt then the folder would be named CSAW_mySampleSheet. This facilitates using multiple sample sheets. Similarly, _MACS2 portion will be different if you use HMMRATAC or Genrich for peak calling.
Note
If you provide a sampleSheet with name, condition and group columns, "multiple comparison mode" will be detected. The original sampleSheet will be split on the group column, and multiple pairwise comparisons will be run with CSAW, one per group.
Note
The output from Genrich will be peaks called per-group if you specify a sample sheet. This is because Genrich is capable of directly using replicates during peak calling.
Where to find final bam files and biwgwigs¶
Bam files with the extention filtered.bam are only filtered for PCR duplicates. The final bam files filtered additionally for fragment size and used as direct input to MACS2 are found in the short_bams folder with the exention .short.cleaned.bam.
Bigwig files calculated from these bam files are found under deepTools_ATAC/bamCompare with the extention .filtered.bw.
Command line options¶
MPI-IE workflow for ATAC-seq Analysis
- usage example:
ATAC-seq -d working-dir mm10
usage: ATAC-seq -d WORKINGDIR [-h] [-v] [-c CONFIGFILE]
[--clusterConfigFile CLUSTERCONFIGFILE] [-j INT] [--local]
[--keepTemp] [--snakemakeOptions SNAKEMAKEOPTIONS] [--DAG]
[--version] [--emailAddress EMAILADDRESS]
[--smtpServer SMTPSERVER] [--smtpPort SMTPPORT] [--onlySSL]
[--emailSender EMAILSENDER] [--smtpUsername SMTPUSERNAME]
[--smtpPassword SMTPPASSWORD]
[--peakCaller {MACS2,HMMRATAC,Genrich}]
[--maxFragmentSize MAXFRAGMENTSIZE]
[--minFragmentSize MINFRAGMENTSIZE] [--qval INT]
[--sampleSheet SAMPLESHEET] [--externalBed EXTERNALBED]
[--fromBAM FROMBAM] [--bamExt BAMEXT] [--FDR FDR]
[--LFC ABSBESTLFC]
GENOME
Positional Arguments¶
- GENOME
Genome acronym of the target organism. Either a yaml file or one of: hg38, mm39_ens106, GRCh38_gencode40, dm6, GRCz10, SchizoSPombe_ASM294v2, mm10_gencodeM19, GRCz11
Required Arguments¶
- -d, --working-dir
working directory is output directory and must contain DNA-mapping pipeline output files
General Arguments¶
- -v, --verbose
verbose output (default: 'False')
- -c, --configFile
configuration file: config.yaml (default: 'None')
- --clusterConfigFile
configuration file for cluster usage. In absence, the default options specified in defaults.yaml and workflows/[workflow]/cluster.yaml would be selected (default: 'None')
- -j, --jobs
maximum number of concurrently submitted Slurm jobs / cores if workflow is run locally (default: '5')
- --local
run workflow locally; default: jobs are submitted to Slurm queue (default: 'False')
- --keepTemp
Prevent snakemake from removing files marked as being temporary (typically intermediate files that are rarely needed by end users). This is mostly useful for debugging problems.
- --snakemakeOptions
Snakemake options to be passed directly to snakemake, e.g. use --snakemakeOptions='--dryrun --rerun-incomplete --unlock --forceall'. WARNING! ONLY EXPERT USERS SHOULD CHANGE THIS! THE DEFAULT VALUE WILL BE APPENDED RATHER THAN OVERWRITTEN! (default: '['--use-conda']')
- --DAG
If specified, a file ending in _pipeline.pdf is produced in the output directory that shows the rules used and their relationship to each other.
- --version
show program's version number and exit
Email Arguments¶
- --emailAddress
If specified, send an email upon completion to the given email address
- --smtpServer
If specified, the email server to use.
- --smtpPort
The port on the SMTP server to connect to. A value of 0 specifies the default port.
- --onlySSL
The SMTP server requires an SSL connection from the beginning.
- --emailSender
The address of the email sender. If not specified, it will be the address indicated by --emailAddress
- --smtpUsername
If your SMTP server requires authentication, this is the username to use.
- --smtpPassword
If your SMTP server requires authentication, this is the password to use.
Options¶
- --peakCaller
Possible choices: MACS2, HMMRATAC, Genrich
The peak caller to use. The default is "MACS2"
- --maxFragmentSize
Maximum size of (typically nucleosomal) fragments for inclusion in the analysis (default: '150')
- --minFragmentSize
Minimum size of (typically nucleosomal) fragments for inclusion in the analysis (default: '0')
- --qval
qvalue threshold for MACS2(default: '0.001')
- --sampleSheet
Invoke differential accessibility analysis by providing information on samples; see 'https://github.com/maxplanck-ie/snakepipes/tree/master/docs/content/sampleSheet.example.tsv' for example. IMPORTANT: The first entry defines which group of samples are control. With this, the order of comparison and likewise the sign of values can be changed! Also, the condition control should not be used (reserved to mark input samples in the ChIP-Seq workflow (default: 'None').
- --externalBed
A bed file with intervals to be tested for differential binding. (default: 'None')
- --fromBAM
Input folder with bam files. If provided, the analysis will start from this point. (default: 'False')
- --bamExt
Extention of provided bam files, will be substracted from basenames to obtain sample names. (default: '"filtered.bam"')
- --FDR
FDR threshold to apply for filtering DB regions(default: '0.05')
- --LFC
Log fold change threshold to apply for filtering DB regions(default: '1')