WGBS
What it does
Optionally trimmed reads are mapped to the reference genome using a bisulfite-specific aligner (bwa-meth). Quality metrics are collected and synthesized in a QC report, including bisulfite conversion rate, mapping rate, coverage metrics, and methylation bias.
There are two flags that allow skipping certain QC metric calculation, i.e. --skipDOC and --GCbias. These deactivate or activate, respectively, the depth of coverage (DOC) calculations or GC bias calculation done by deepTools. If you run the workflow with --fromBAM, you can also choose to skip the re-calculation of most QC metrics with --skipBamQC.
Methylation ratios are extracted (via MethylDackel) for CpG positions in the reference genome with a minimum coverage specified by --minCoverage and low SNP allelic frequency (<0.25 illegitimate bases).
If a sample sheet is provided, Metilene , DMRseq and/or DSS (as specified by --DMRprograms) will be used to find differentially methylated regions (DMRs). Filtering criterion can be changed both for the CpGs used to find DMRs as well as what are considered as significant DMRs.
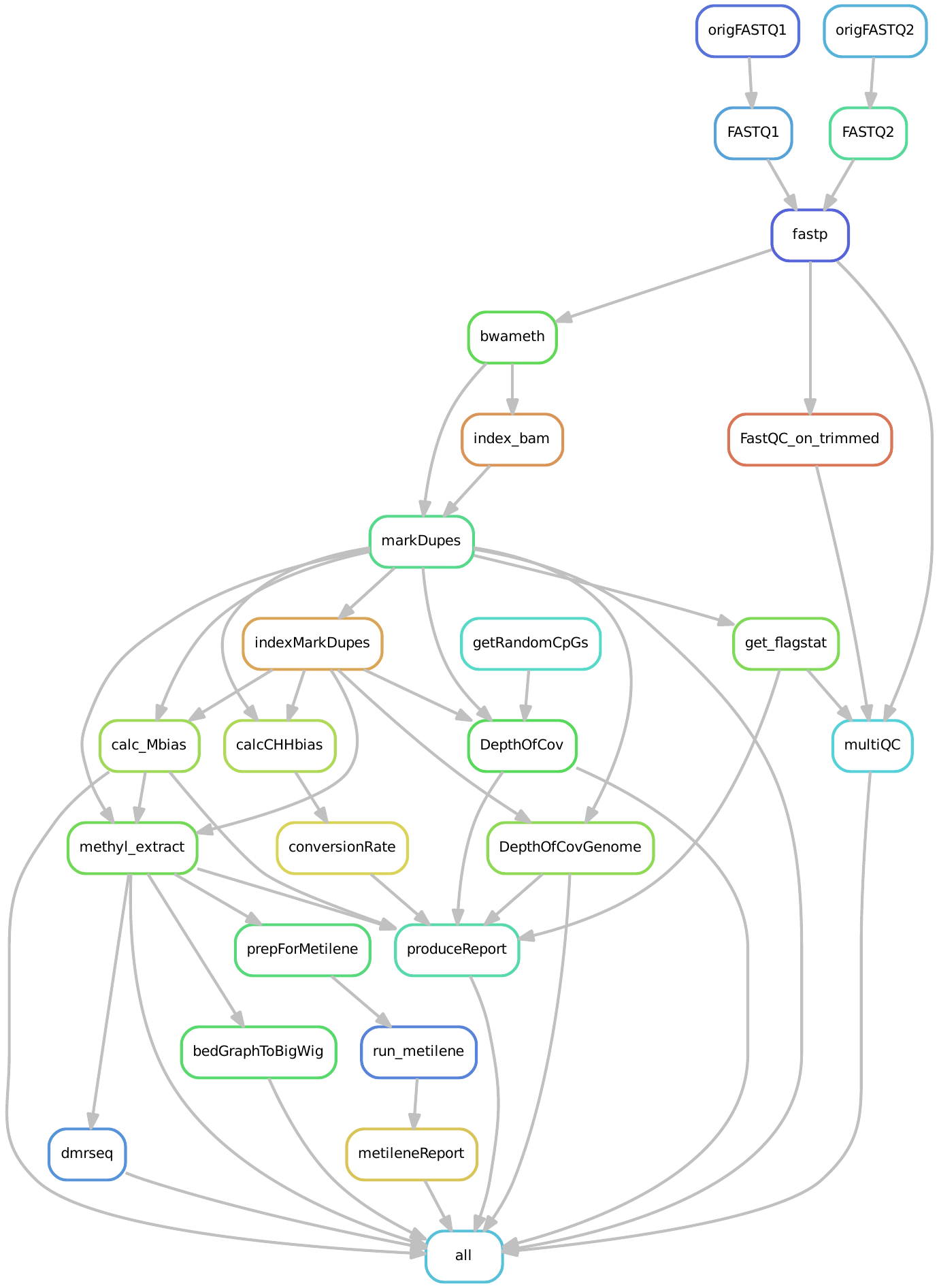
Input requirements
This pipeline requires fastq files and a genome alias, for which bwa-meth index has been built.
Optional inputs include a sample sheet with grouping information to use in differential methylation analysis and a blacklist bed file with genomic positions corresponding to known snps to mask single CpG methylation values.
It is possible to use pipeline-compatible bam files as input. For that, the user has to use the --fromBAM flag and provide the bam file extension if not matched by the default.
Workflow configuration file
## General/Snakemake parameters, only used/set by wrapper or in Snakemake cmdl, but not in Snakefile
outdir:
configFile:
clusterConfigFile:
local: False
maxJobs: 12
## directory with fastq or bam files
indir:
## Genome information
genome:
###SNP black list (bed file)
blacklist:
###sample Sheet
sampleSheet:
###inclusion bounds for methylation extraction
noAutoMethylationBias: False
## FASTQ file extension (default: ".fastq.gz")
ext: '.fastq.gz'
## paired-end read name extension (default: ['_R1', "_R2"])
reads: [_R1, _R2]
## Number of reads to downsample from each FASTQ file
downsample:
## Options for trimming
trim: False
trimmer: 'fastp'
trimmerOptions: '-q 5 -l 30 -M 5'
## Bin size of output files in bigWig format
bwBinSize: 25
## Run FASTQC read quality control
fastqc: false
verbose: False
plotFormat: 'png'
#### Flag to control the pipeline entry point
fromBAM: False
bamExt: '.bam'
pairedEnd: True
###Flags to control skipping of certain QC calculations
skipDOC: False
GCbias: False
###Thresholds for filtering of statistical comparisons (DMRs and DMLs)
DMRprograms: 'metilene,dmrseq'
maxDist: 300
minCpGs: 10
minCoverage: 5
FDR: 0.1
minMethDiff: 0.1
###MethylDackel options
MethylDackelOptions: '--mergeContext --maxVariantFrac 0.25 --minDepth 4'
##umi_tools
UMIBarcode: False
bcPattern: NNNNCCCCCCCCC #default: 4 base umi barcode, 9 base cell barcode (eg. RELACS barcode)
UMIDedup: False
UMIDedupSep: "_"
UMIDedupOpts: --paired
aligner: bwameth
Understanding the outputs
The WGBS pipeline invoked fastq files and a sample sheet and the --trim and --fastqc options will generate the following output:
output_dir
├── bwameth
├── cluster_logs
├── dmrseq_sampleSheet_minCoverage5
├── FASTQ
├── FastQC_trimmed
├── FASTQ_fastp
├── MethylDackel
├── metilene_sampleSheet_minCoverage5
├── multiQC
├── originalFASTQ
└── QC_metrics
The workflow produces the following outputs:
FASTQ_downsampled: contains read files downsampled to 5mln reads. These are used to calculate conversion rate which would otherwise take a very long time.
bwameth: contains bam files obtained through read alignment with bwa-meth and the PCR duplicate removal with sambamba, as well as matching bam index files.
dmrseq_sampleSheet_minCoverage<X>: DMRs (DMRs.txt) and a report (Stats_report.html) from DMRseq as well as a saved R session (Session.RData) using the requested minimum coverage. If you rerun the pipeline with a different minimum coverage specified then a new directory will be created.
DSS_sampleSheet_minCoverage<X>: As with DMRseq above.
FastQC_trimmed: FastQC output on the trimmed reads.
FASTQ_fastp: The trimmed reads and QC metrics from FastP.
MethylDackel: BigWig coverage and methylation files as well as the bedGraph files produced by MethylDackel.
metilene_sampleSheet_minCoverage<X>: contains output files from metilene in DMRs.txt. DMRs.annotated.txt is an annotated version of that, wherein DMRs are annotated with the nearest gene and the distance to it. There is additionally a QC report (Stats_report.html) that summarizes various properties of the DMRs.
QC_metrics: contains output files from conversion rate, flagstat, depth of coverage, GCbias and methylation bias calculations. The QC report in pdf format collecting those metrics in tabular form is also found in this folder.
Example output plots
Using data from Habibi et al., Cell Stem Cell 2013 corresponding to mouse chr6:4000000-6000000, following plots could be obtained:
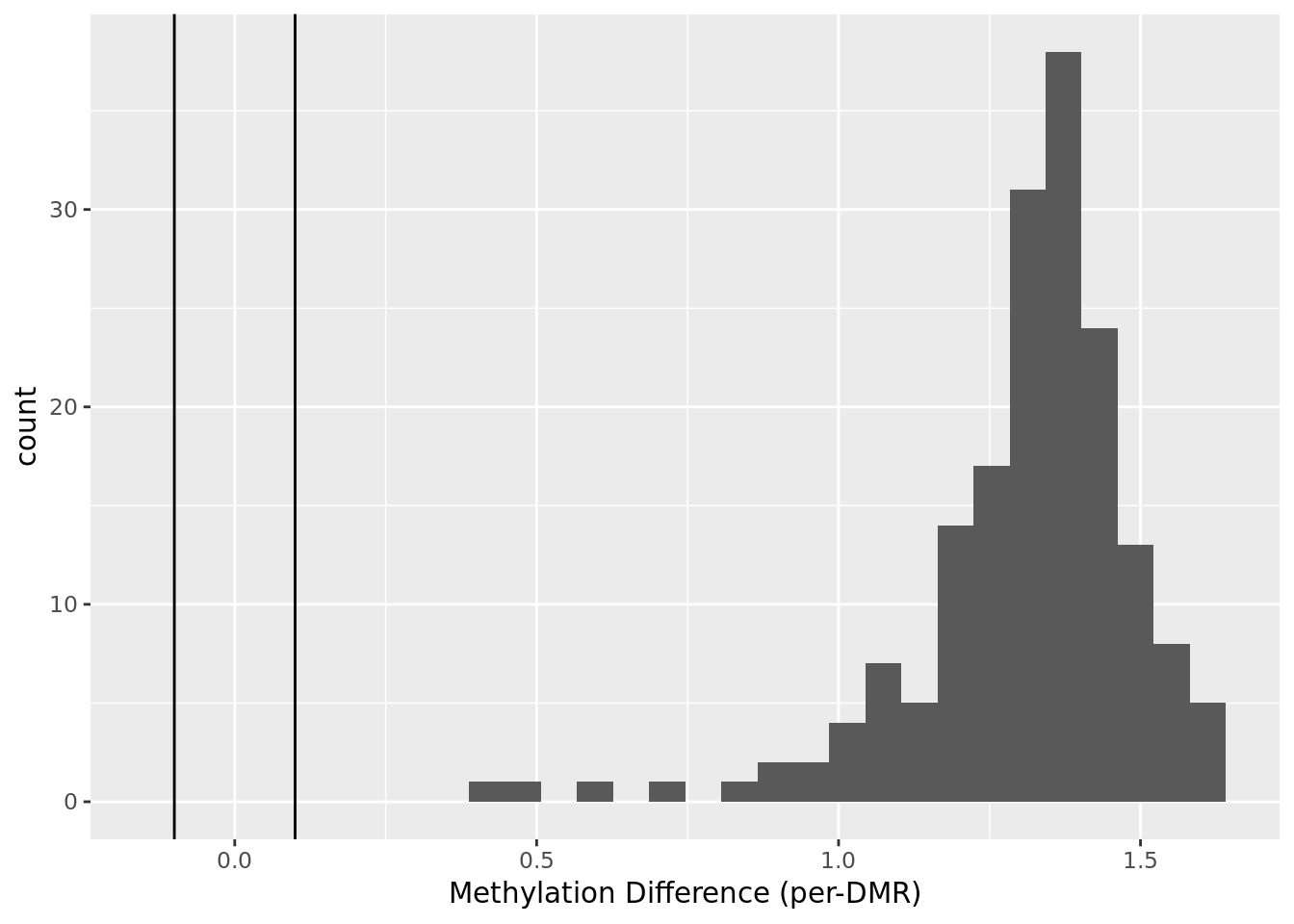
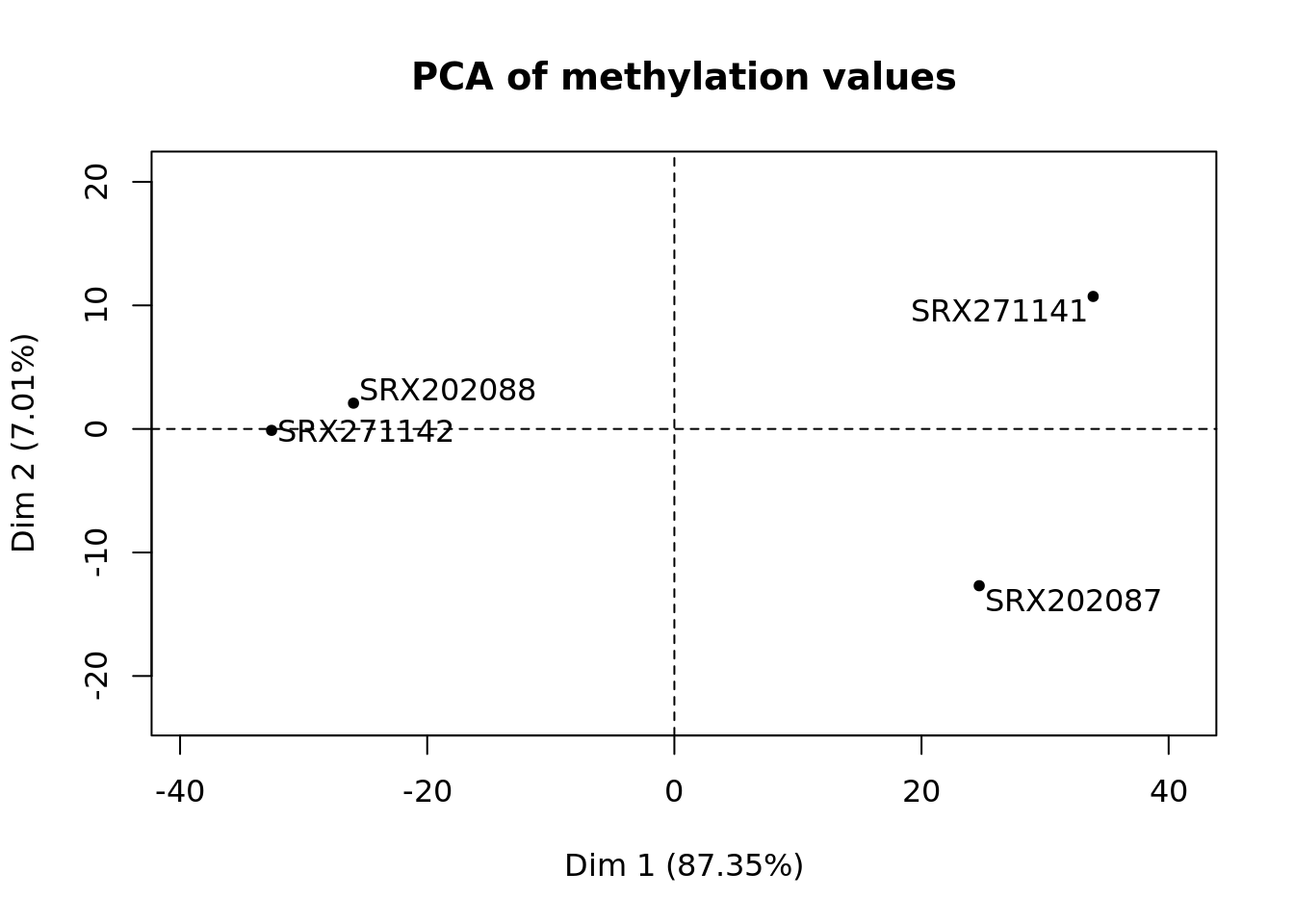
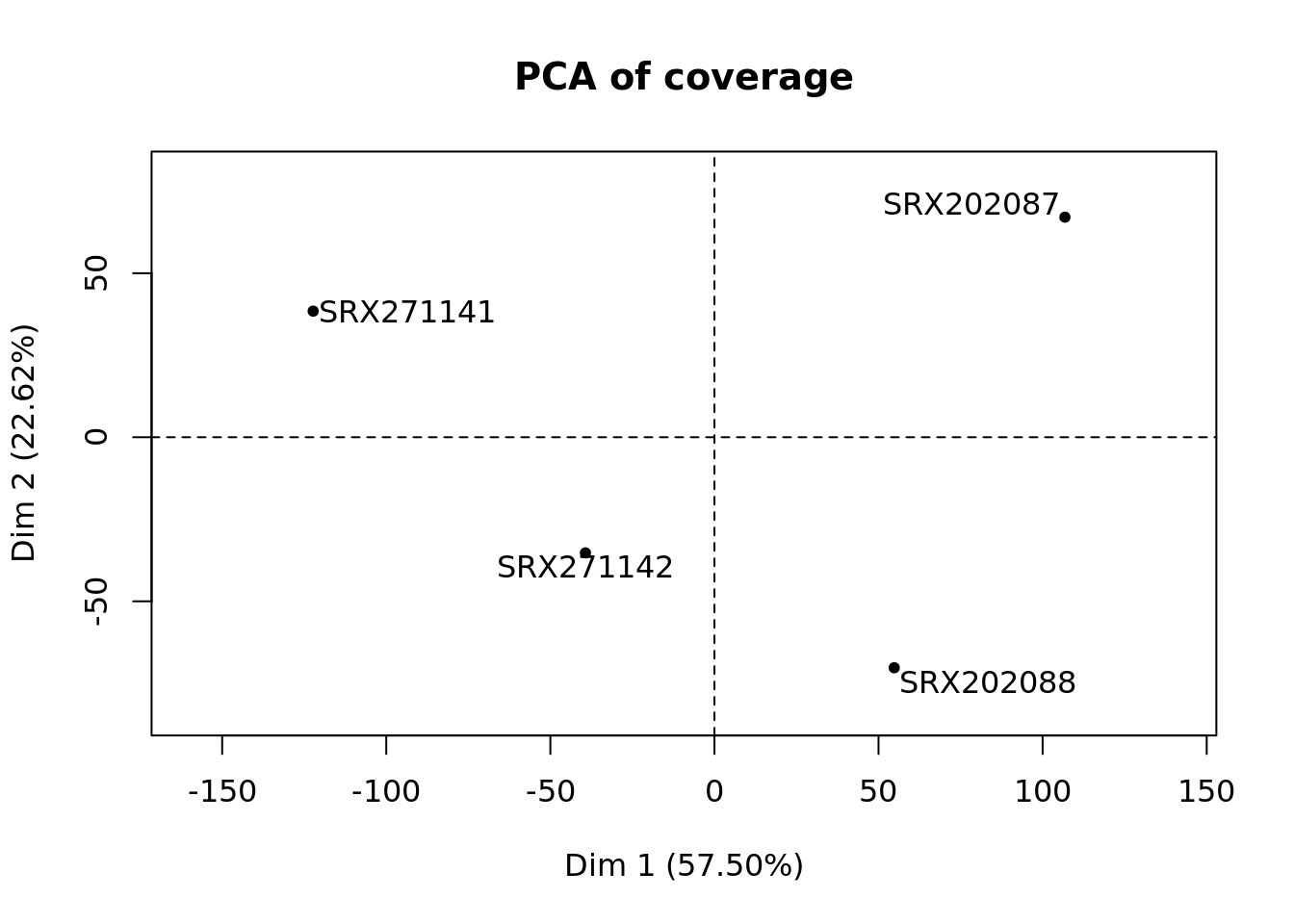
Command line options
MPI-IE workflow for WGBS analysis
- usage example:
WGBS -i read_input_dir -o output-dir mm10
usage: WGBS -i INDIR -o OUTDIR [-h] [-v] [--ext EXT] [--reads READS READS]
[-c CONFIGFILE] [--keepTemp] [--snakemakeOptions SNAKEMAKEOPTIONS]
[--DAG] [--version] [--emailAddress EMAILADDRESS]
[--smtpServer SMTPSERVER] [--smtpPort SMTPPORT] [--onlySSL]
[--emailSender EMAILSENDER] [--smtpUsername SMTPUSERNAME]
[--smtpPassword SMTPPASSWORD] [--downsample INT] [--trim]
[--trimmer {cutadapt,trimgalore,fastp}]
[--trimmerOptions TRIMMEROPTIONS] [--fastqc] [--bcExtract]
[--bcPattern BCPATTERN] [--UMIDedup] [--UMIDedupSep UMIDEDUPSEP]
[--UMIDedupOpts UMIDEDUPOPTS] [--bwBinSize BWBINSIZE]
[--plotFormat STR] [--aligner {bwameth,bwameth2}]
[--blacklist BLACKLIST] [--targetRegions TARGETREGIONS]
[--sampleSheet SAMPLESHEET] [--noAutoMethylationBias]
[--maxDist MAXDIST] [--minCpGs MINCPGS]
[--minMethDiff MINMETHDIFF] [--minCoverage MINCOVERAGE]
[--FDR FDR] [--MethylDackelOptions METHYLDACKELOPTIONS]
[--fromBAM] [--skipBamQC] [--bamExt BAMEXT] [--singleEnd]
[--DMRprograms DMRPROGRAMS] [--metileneOptions METILENEOPTIONS]
[--skipDOC] [--GCbias]
GENOME
Positional Arguments
- GENOME
Genome acronym of the target organism. Either a yaml file or one of: mm39_ens106, GRCz11, SchizoSPombe_ASM294v2, GRCh38_gencode40, hg38, GRCz10, mm10_gencodeM19, dm6
Required Arguments
- -i, --input-dir
input directory containing the FASTQ files, either paired-end OR single-end data
- -o, --output-dir
output directory
General Arguments
- -v, --verbose
verbose output (default: 'False')
- --ext
Suffix used by input fastq files (default: ''.fastq.gz'').
- --reads
Suffix used to denote reads 1 and 2 for paired-end data. This should typically be either '_1' '_2' or '_R1' '_R2' (default: '['_R1', '_R2']). Note that you should NOT separate the values by a comma (use a space) or enclose them in brackets.
- -c, --configFile
configuration file: config.yaml (default: 'None')
- --keepTemp
Prevent snakemake from removing files marked as being temporary (typically intermediate files that are rarely needed by end users). This is mostly useful for debugging problems.
- --snakemakeOptions
Snakemake options to be passed directly to snakemake, e.g. use --snakemakeOptions='--dryrun --rerun-incomplete --unlock --forceall'. WARNING! ONLY EXPERT USERS SHOULD CHANGE THIS! THE DEFAULT VALUE WILL BE APPENDED RATHER THAN OVERWRITTEN! (default: '['--use-conda']')
- --DAG
If specified, a file ending in _pipeline.pdf is produced in the output directory that shows the rules used and their relationship to each other.
- --version
show program's version number and exit
Email Arguments
- --emailAddress
If specified, send an email upon completion to the given email address
- --smtpServer
If specified, the email server to use.
- --smtpPort
The port on the SMTP server to connect to. A value of 0 specifies the default port.
- --onlySSL
The SMTP server requires an SSL connection from the beginning.
- --emailSender
The address of the email sender. If not specified, it will be the address indicated by --emailAddress
- --smtpUsername
If your SMTP server requires authentication, this is the username to use.
- --smtpPassword
If your SMTP server requires authentication, this is the password to use.
Options
- --downsample
Downsample the given number of reads randomly from of each FASTQ file (default: 'None')
- --trim
Activate fastq read trimming. If activated, Illumina adaptors are trimmed by default. Additional parameters can be specified under --trimmerOptions. (default: 'True')
- --trimmer
Possible choices: cutadapt, trimgalore, fastp
Trimming program to use: Cutadapt, TrimGalore, or fastp. Note that if you change this you may need to change --trimmerOptions to match! (default: ''fastp'')
- --trimmerOptions
Additional option string for trimming program of choice. (default: ''-q 5 -l 30 -M 5'')
- --fastqc
Run FastQC read quality control (default: 'False')
- --bcExtract
To extract umi barcode from fastq file via UMI-tools and add it to the read name (default: 'False')
- --bcPattern
The pattern to be considered for the barcode. 'N' = UMI position (required) 'C' = barcode position (optional) (default: '''')
- --UMIDedup
Deduplicate bam file based on UMIs via umi_tools dedup that are present in the read name. (default: 'False')
- --UMIDedupSep
umi separation character that will be passed to umi_tools.(default: ''_'')
- --UMIDedupOpts
Additional options that will be passed to umi_tools.(default: '''')
- --bwBinSize
Bin size of output files in bigWig format (default: '25')
- --plotFormat
Possible choices: png, pdf, None
Format of the output plots from deepTools. Select 'none' for no plots (default: ''png'')
- --aligner
Possible choices: bwameth, bwameth2
Program used for mapping: bwameth or bwameth2 (default: 'None'). Specifying bwameth2 will run bwameth with bwa-mem2 underneath.
- --blacklist
Bed file(s) with positions to mask for methylation calling. Useful for masking SNPs in your strain of interest. (default: 'None')
- --targetRegions
Bed file(s) with regions of interest to evaluate methylation for. (default: 'None')
- --sampleSheet
Perform differential methylation analysis between groups of samples by providing a text file with sample information to use for statistical analysis. (default: 'None')
- --noAutoMethylationBias
If specified, MethylDackel mbias will NOT be run and the suggested parameters from it will NOT be used for methylation extraction. You can instead supply them manually in --MethylDackelOptions.
- --maxDist
The maximum distance between CpGs in a DMR (for metilene, default: '300')
- --minCpGs
The minimum number of CpGs in a DMR (for metilene, default: '10')
- --minMethDiff
The minimum methylation change in methylation for CpG inclusion in DMR detection (for metilene, default: '0.1')
- --minCoverage
The minimum coverage needed for across all samples for a CpG to be used in DMR calling and PCA. Note that you can change this value without overwritting the DMR output. Default: '5'
- --FDR
FDR threshold for returned DMRs (default: '0.1')
- --MethylDackelOptions
Options to pass to MethylDackel extract. You are highly advised NOT to set a minimum coverage at this step. Default: ''--mergeContext --maxVariantFrac 0.25 --minDepth 4''
- --fromBAM
If specified, the input is taking from BAM files containing alignments rather than fastq files. See also --bamExt.
- --skipBamQC
If specified, filtering of input bam files, as well as calculation of quality metrics, will be skipped.
- --bamExt
If --fromBAM is specified, this is the expected file extension. Removing it yields sample names. Default: ''.bam''
- --singleEnd
If --fromBAM is specified, this indicates that the input BAM files contain paired-end data. The option is ignored unless --fromBAM is given.
- --DMRprograms
If a sample sheet is provided, use the specified DMR-calling programs. Multiple programs can be comma-separated with no spaces (e.g., 'metilene,dmrseq,DSS'). The available programs are metilene, dmrseq, and DSS (note that this is very slow). Default: 'metilene,dmrseq'.
- --metileneOptions
Options to pass to metilene. Default: 'None'
- --skipDOC
Skip depth of coverage calculation with deepTools.
- --GCbias
Perform GC bias calculation with deepTools.