DNA-mapping¶
What it does¶
This is the primary DNA-mapping pipeline. It can be used both alone or upstream of the ATAC-seq and ChIP-seq pipelines. This has a wide array of options, including trimming and various QC steps (e.g., marking duplicates and plotting coverage and PCAs). In addition, basic coverage tracks are created to facilitate viewing the data in IGV.
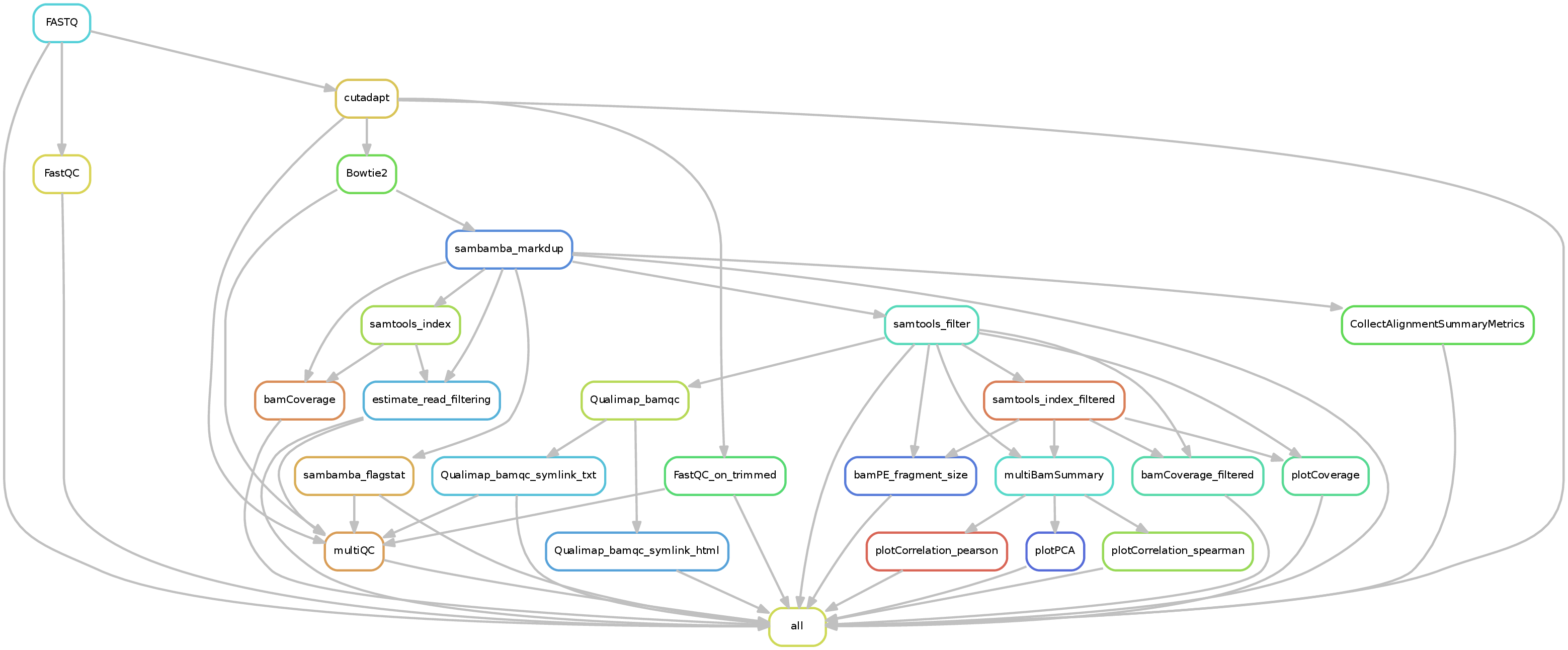
Input requirements¶
The only requirement is a directory of gzipped fastq files. Files could be single or paired end, and the read extensions could be modified using the keys in the defaults.yaml file below.
Configuration file¶
There is a configuration file in snakePipes/workflows/DNA-mapping/defaults.yaml:
## General/Snakemake parameters, only used/set by wrapper or in Snakemake cmdl, but not in Snakefile
pipeline: dna-mapping
outdir:
configFile:
clusterConfigFile:
local: False
maxJobs: 5
## directory with fastq files
indir:
## preconfigured target genomes (mm9,mm10,dm3,...) , see /path/to/snakemake_workflows/shared/organisms/
## Value can be also path to your own genome config file!
genome:
## FASTQ file extension (default: ".fastq.gz")
ext: '.fastq.gz'
## paired-end read name extension (default: ['_R1', "_R2"])
reads: [_R1, _R2]
## mapping mode
mode: mapping
aligner: Bowtie2
## Number of reads to downsample from each FASTQ file
downsample:
## Options for trimming
trim: False
trimmer: cutadapt
trimmerOptions:
## Bin size of output files in bigWig format
bwBinSize: 25
## Run FASTQC read quality control
fastqc: false
## Run computeGCBias quality control
GCBias: false
## Retain only de-duplicated reads/read pairs
dedup: false
## Retain only reads with at least the given mapping quality
mapq: 0
## Retain only reads mapping in proper pairs
properPairs: false
## Mate orientation in paired-end experiments for Bowtie2 mapping
## (default "--fr" is appropriate for Illumina sequencing)
mateOrientation: --fr
## other Bowtie2 stuff
insertSizeMax: 1000
alignerOpts:
plotFormat: png
UMIBarcode: False
bcPattern: NNNNCCCCCCCC #default: 4 base umi barcode, 8 base cell barcode (eg. RELACS barcode)
UMIDedup: False
UMIDedupSep: "_"
UMIDedupOpts:
## Median/mean fragment length, only relevant for single-end data (default: 200)
fragmentLength: 200
qualimap: false
verbose: false
Many of these options can be more conveniently set on the command-line (e.g., --qualimap sets qualimap: true). However, you may need to change the reads: setting if your paired-end files are not denoted by sample_R1.fastq.gz and sample_R2.fastq.gz, but rather sample_1.fastq.gz and sample_2.fastq.gz.
Understanding the outputs¶
The DNA mapping pipeline will generate output of the following structure:
.
├── bamCoverage
├── Bowtie2
├── deepTools_qc
│ ├── bamPEFragmentSize
│ ├── estimateReadFiltering
│ ├── multiBamSummary
│ ├── plotCorrelation
│ ├── plotCoverage
│ └── plotPCA
├── FASTQ
├── FastQC
├── filtered_bam
├── multiQC
│ └── multiqc_data
└── Sambamba
In addition to the FASTQ module results (see Running snakePipes), the workflow produces the following outputs:
Bowtie2 : Contains the BAM files after mapping with Bowtie2 and indexed by Samtools.
filtered_bam : Contains the BAM files filtered by the provided criteria, such as mapping quality (
--mapq) or PCR duplicates (--dedup). This file is used for most downstream analysis in the DNA-mapping and ChIP-seq/ATAC-seq pipeline.bamCoverage : Contains the coverage files (bigWig format) produced from the BAM files by deepTools bamCoverage . The files are either raw, or 1x normalized (by sequencing depth). They are useful for plotting and inspecting the data in IGV.
deepTools_qc : Contains various QC files and plots produced by deepTools on the filtered BAM files. These are very useful for evaluation of data quality. The folders are named after the tools. Please look at the deepTools documentation on how to interpret the outputs from each tool.
Sambamba : Contains the alignment metrices evaluated on the BAM files by Sambamba.
A number of other directories may optionally be present if you specified read trimming, using Qualimap, or a variety of other options. These are typically self-explanatory.
A fair number of useful QC plots are or can be generated by the pipeline. These include correlation and PCA plots as well as the output from MultiQC.
Command line options¶
MPI-IE workflow for DNA mapping
- usage example:
DNA-mapping -i input-dir -o output-dir mm10
usage: DNA-mapping -i INDIR -o OUTDIR [-h] [-v] [--ext EXT]
[--reads READS READS] [-c CONFIGFILE]
[--clusterConfigFile CLUSTERCONFIGFILE] [-j INT] [--local]
[--keepTemp] [--snakemakeOptions SNAKEMAKEOPTIONS] [--DAG]
[--version] [--emailAddress EMAILADDRESS]
[--smtpServer SMTPSERVER] [--smtpPort SMTPPORT] [--onlySSL]
[--emailSender EMAILSENDER] [--smtpUsername SMTPUSERNAME]
[--smtpPassword SMTPPASSWORD] [--VCFfile VCFFILE]
[--strains STRAINS] [--SNPfile SNPFILE]
[--NMaskedIndex NMASKEDINDEX] [-m MODE] [--downsample INT]
[--trim] [--trimmer {cutadapt,trimgalore,fastp}]
[--trimmerOptions TRIMMEROPTIONS] [--fastqc] [--bcExtract]
[--bcPattern BCPATTERN] [--UMIDedup]
[--UMIDedupSep UMIDEDUPSEP] [--UMIDedupOpts UMIDEDUPOPTS]
[--bwBinSize BWBINSIZE] [--plotFormat STR]
[--alignerOpts ALIGNEROPTS] [--cutntag]
[--mateOrientation MATEORIENTATION] [--qualimap] [--dedup]
[--properPairs] [--mapq INT]
[--insertSizeMax INSERTSIZEMAX] [--GCBias]
[--aligner {Bowtie2,bwa,bwa-mem2}]
GENOME
Positional Arguments¶
- GENOME
Genome acronym of the target organism. Either a yaml file or one of: hg38, mm39_ens106, GRCh38_gencode40, dm6, GRCz10, SchizoSPombe_ASM294v2, mm10_gencodeM19, GRCz11
Required Arguments¶
- -i, --input-dir
input directory containing the FASTQ files, either paired-end OR single-end data
- -o, --output-dir
output directory
General Arguments¶
- -v, --verbose
verbose output (default: 'False')
- --ext
Suffix used by input fastq files (default: '".fastq.gz"').
- --reads
Suffix used to denote reads 1 and 2 for paired-end data. This should typically be either '_1' '_2' or '_R1' '_R2' (default: '['_R1', '_R2']). Note that you should NOT separate the values by a comma (use a space) or enclose them in brackets.
- -c, --configFile
configuration file: config.yaml (default: 'None')
- --clusterConfigFile
configuration file for cluster usage. In absence, the default options specified in defaults.yaml and workflows/[workflow]/cluster.yaml would be selected (default: 'None')
- -j, --jobs
maximum number of concurrently submitted Slurm jobs / cores if workflow is run locally (default: '5')
- --local
run workflow locally; default: jobs are submitted to Slurm queue (default: 'False')
- --keepTemp
Prevent snakemake from removing files marked as being temporary (typically intermediate files that are rarely needed by end users). This is mostly useful for debugging problems.
- --snakemakeOptions
Snakemake options to be passed directly to snakemake, e.g. use --snakemakeOptions='--dryrun --rerun-incomplete --unlock --forceall'. WARNING! ONLY EXPERT USERS SHOULD CHANGE THIS! THE DEFAULT VALUE WILL BE APPENDED RATHER THAN OVERWRITTEN! (default: '['--use-conda']')
- --DAG
If specified, a file ending in _pipeline.pdf is produced in the output directory that shows the rules used and their relationship to each other.
- --version
show program's version number and exit
Email Arguments¶
- --emailAddress
If specified, send an email upon completion to the given email address
- --smtpServer
If specified, the email server to use.
- --smtpPort
The port on the SMTP server to connect to. A value of 0 specifies the default port.
- --onlySSL
The SMTP server requires an SSL connection from the beginning.
- --emailSender
The address of the email sender. If not specified, it will be the address indicated by --emailAddress
- --smtpUsername
If your SMTP server requires authentication, this is the username to use.
- --smtpPassword
If your SMTP server requires authentication, this is the password to use.
Allele-specific mapping arguments¶
- --VCFfile
VCF file to create N-masked genomes (default: 'None')
- --strains
Name or ID of SNP strains separated by comma (default: 'None')
- --SNPfile
File containing SNP locations (default: 'None')
- --NMaskedIndex
N-masked index of the reference genome (default: 'None'). Note that this should point to a file (i.e. 'Genome' for STAR indices, genome.1.bt2 for bowtie2 indices).
Options¶
- -m, --mode
workflow running modes (available: 'mapping,allelic-mapping')(default: '"mapping"')
- --downsample
Downsample the given number of reads randomly from of each FASTQ file (default: 'False')
- --trim
Activate fastq read trimming. If activated, Illumina adaptors are trimmed by default. Additional parameters can be specified under --trimmerOptions. (default: 'False')
- --trimmer
Possible choices: cutadapt, trimgalore, fastp
Trimming program to use: Cutadapt, TrimGalore, or fastp. Note that if you change this you may need to change --trimmerOptions to match! (default: '"cutadapt"')
- --trimmerOptions
Additional option string for trimming program of choice. (default: '')
- --fastqc
Run FastQC read quality control (default: 'False')
- --bcExtract
To extract umi barcode from fastq file via UMI-tools and add it to the read name (default: 'False')
- --bcPattern
The pattern to be considered for the barcode. 'N' = UMI position (required) 'C' = barcode position (optional) (default: '"NNNNCCCCCCCC"')
- --UMIDedup
Deduplicate bam file based on UMIs via umi_tools dedup that are present in the read name. (default: 'False')
- --UMIDedupSep
umi separation character that will be passed to umi_tools.(default: '"_"')
- --UMIDedupOpts
Additional options that will be passed to umi_tools.(default: '')
- --bwBinSize
Bin size of output files in bigWig format (default: '25')
- --plotFormat
Possible choices: png, pdf, None
Format of the output plots from deepTools. Select 'none' for no plots (default: '"png"')
- --alignerOpts
Options that will be passed to Bowtie2 or bwa. You can specify things such as --local or --very-sensitive here. The mate orientation and maximum insert size are specified elsewhere. Read group information is set automatically. Note that you may need to escape the first - (e.g., '--very-fast'). Default: ''.
- --cutntag
if set, Bowti2 is used for mapping with parameters as has been used in the method section of Kaya-okur et al. 2019. ('--local --very-sensitive-local --no-mixed --no-discordant --phred33 -I 10 -X 700')Setting this flag overwrites the '--alignerOpts' and '--insertSizeMax'. Default is 'False'.
- --mateOrientation
The --fr, --ff, or --rf option for bowtie2 (default: '"--fr"')
- --qualimap
activate Qualimap (default: 'False')
- --dedup
retain only de-duplicated reads/read pairs (given single-/paired-end data), recommended for ChIP-seq data (default: 'False')
- --properPairs
retain only reads mapping in proper pairs (default: 'False')
- --mapq
retain only reads with at least the given mapping quality. We recommend usingmapq of 3 or more for ChIP-seq to remove all true multimapping reads. (default: '0')
- --insertSizeMax
Maximum insert size allowed during mapping (default: '1000')
- --GCBias
run computeGCBias quality control (long runtime!). Note that GCBias analysis is skipped if downsampling is specified (default: 'False')
- --aligner
Possible choices: Bowtie2, bwa, bwa-mem2
Program used for mapping: Bowtie2 or bwa (default: '"Bowtie2"').